
Anlage 2 Bek GS 910 - Leitfaden zur Quantifizierung von Krebsrisikozahlen bei Exposition gegenüber krebserzeugenden Gefahrstoffen für die Grenzwertsetzung am Arbeitsplatz (1)
Anlage 2 zu Bekanntmachung zu Gefahrstoffen 910
1 Rahmen der Risikoquantifizierung
1.1 Vorbemerkung: Prinzipien der Risikoquantifizierung bei begrenzter Datenbasis
(1) Der vorliegende Leitfaden soll die Voraussetzungen schaffen, um Expositions-Risiko-Beziehungen für krebserzeugende Stoffe nach harmonisierten Regeln zu beschreiben, und dabei die Option einschließen, Arbeitsplatzgrenzwerte für diese Stoffe zu begründen. Dazu werden Kriterien aufgestellt, um die Eignung vorliegender Daten zu einem Stoff zu bewerten, und Vorgehensweisen empfohlen, aus diesen Daten bestmöglich Expositions-Risiko-Beziehungen zu ermitteln.
(2) Der Schutz von Beschäftigten am Arbeitsplatz gegenüber krebserzeugenden Chemikalien (Kanzerogene, Karzinogene) wird insbesondere durch die EU-Richtlinie 2004/37/EG (Krebsrichtlinie; EU, 2004) und durch die Gefahrstoffverordnung (GefStoffV; Bundesministerium für Arbeit und Soziales, 2005) geregelt. Im Sinne der Krebsrichtlinie bezeichnet "Karzinogen" einen Stoff, der die in Anhang VI der Richtlinie 67/548/EWG (EU, 2007) genannten Kriterien für die Einstufung als krebserzeugender Stoff der Kategorie 1 oder 2 erfüllt. Stoffe der Kategorien 1 und 2 für krebserzeugende Stoffe ("Karzinogene") sind sowohl im Sinne der Krebsrichtlinie als auch nach der GefStoffV im Risikomanagement gleich zu behandeln. Es ist gemäß diesen Bestimmungen also unerheblich, ob ein Stoff aufgrund epidemiologischer Erkenntnisse (Kategorie 1) oder aufgrund von Tierversuchen (Kategorie 2) als krebserzeugend erkannt und eingestuft wurde (5). Da eine Krebserkrankung als eine besonders schwere Erkrankung anzusehen ist und da die Krebsrichtlinie davon ausgeht, dass ein Expositionsniveau, unterhalb dessen eine Gefährdung der Gesundheit nicht mehr gegeben ist, nicht festgelegt werden kann, sehen die rechtlichen Bestimmungen besonders weitgehende Schutzmaßnahmen für diese Stoffe vor.
(3) Wegen ihres unmittelbaren Bezugs zum Menschen haben Daten aus epidemiologischen Studien oder aus Studien am Menschen gegenüber den Daten aus Tierversuchen zur Beschreibung von Expositions-Risiko-Beziehungen ein besonderes Gewicht. Allerdings stellen solche Human-Daten mit einer möglicherweise besseren Datenqualität einen nicht wünschenswerten Ausnahmefall dar (da in diesem Falle auch Effekte am Menschen aufgetreten sein müssen), so dass die für die letztendliche Bewertung verbleibende höhere Unsicherheit mit Daten aus dem Tierexperiment in der Regel bewusst in Kauf zu nehmen ist. Unsicherheiten in der Epidemiologie bestehen bei der Abschätzung der Exposition, da in der Regel Messwerte für historische Belastungen fehlen und personenbezogene Expositions-Abschätzungen ungenau sind. Darüber hinaus ist bei epidemiologischen Beobachtungsstudien (nicht-interventionellen Studien) immer der mögliche Einfluss von unkontrollierten Störgrößen zu diskutieren. Tierexperimentelle Daten können dagegen unter kontrollierten Bedingungen und gut definierten Expositionsverhältnissen erhoben werden mit dem Nachteil, dass Tierexperimente nicht mit vergleichbar großen Fallzahlen wie epidemiologische Studien angelegt werden. Die daraus folgenden jeweiligen Einschränkungen in der statistischen Belastbarkeit der gefundenen Dosis-Wirkungs-Beziehung sollten entsprechend beachtet werden. Bei der Übertragung tierexperimenteller Befunde müssen außerdem die Speziesunterschiede in Hinblick auf Dosisäquivalente und Wirkungsmechanismen berücksichtigt werden.
(4) Die Frage der Regulation für krebserzeugende Gefahrstoffe stellt sich jedoch unabhängig von der Eignung der Datenbasis. Das Risikomanagement muss dabei mit den vorhandenen, oft nicht ausreichend belastbaren Expositions-Risiko-Beziehungen einen Grenzwert festlegen. Daher sollten die Unsicherheiten für jede getroffene Entscheidung ermittelt und ausgewiesen werden. Selbst die Schlussfolgerung, dass die Datengrundlage nicht ausreicht, um eine quantitative Expositions-Risiko-Beziehung aufzustellen, ist möglich. Kenntnisse über die Wirkungsmechanismen können in die gewählte Expositionsmetrik und in die Bewertung der Form der beobachteten Expositions-Risiko-Beziehung einfließen. Die möglichen Wirkungsmechanismen sollten bei der Risiko-Extrapolation berücksichtigt werden. Im Ergebnis liegt eine Reihe von Bewertungsmaßstäben mit unterschiedlicher Sicherheit der Extrapolation vor.
(5) In den Fachwissenschaften werden neuerdings auch Mindestdosen (so genannte Wirkungsschwellen) für krebserzeugende Stoffe diskutiert, d.h. Expositionsbereiche, unterhalb derer - z. B. aufgrund der wirksamen biologischen Schutz- und Reparaturmechanismen - eine Gefährdung entgegen bisheriger Überzeugung als unwahrscheinlich gelten. Dies ist jedoch umstritten, außerdem sind der Beweis und die Ermittlung solcher Schwellen methodisch problematisch (Lutz, 2000; Neumann, 2006a, b, c). Nur bei hinreichender Absicherung, die über Plausibilitätsüberlegungen hinaus (z. B. über den angenommenen Wirkungsmechanismus) auch eine quantitative Eingrenzung beinhalten sollte, bei welcher Expositionshöhe diese Wirkungsschwellen anzusiedeln sind, sind solche Erkenntnisse derzeit regulatorisch umsetzbar. Der quantitativen Risikoabschätzung in Verbindung mit Konventionen über Risikoakzeptanz kommt daher besondere Bedeutung bei der Festlegung von Grenzwerten für krebserzeugende Stoffe zu. Unter dem "Risiko" ist dabei das über das Hintergrundrisiko hinausgehende absolute Lebenszeitrisiko bei einer bestimmten Exposition zu verstehen (genauere Definition: siehe Nummer 1.4 sowie Glossar).
(6) Für das Verständnis von Risikobewertungen nach dem vorliegenden Leitfaden ist es wichtig, die Rahmenbedingungen und wissenschaftlichen Grenzen zu kennen, diese auszuweisen und die unter der gegebenen Datenlage getroffene Bewertung bis zur Schaffung einer besseren Datenlage zu akzeptieren. Während derzeit von wissenschaftlicher Seite weder ein "wahres" Risiko und daher auch kein "wahrer" Grenzwert ermittelt werden kann, muss das Risikomanagement die wissenschaftliche Bewertung als derzeit bestmögliche Ableitung und somit als "vermutlich wahr" annehmen, um handlungsfähig zu sein. Da Expositions-Risiko-Beziehungen und Grenzwerte als vorweggenommene Gutachten und im Sinne der Vorsorge abgeleitet werden, ist diese Annahme, nicht zuletzt auch rechtlich, möglich.
(7) Der vorliegende Leitfaden befasst sich mit den wissenschaftlich-methodischen Konventionen, die zur Überbrückung der Kenntnislücken im Bereich akzeptabler und tolerabler Expositionen gegenüber krebserzeugenden Stoffen verwendet werden sollen. Die Abwägung wirtschaftlicher Interessen und des gesellschaftlichen Nutzens von Technologien gegenüber gesundheitlichen Risiken von Beschäftigten ist nicht Gegenstand dieses Leitfadens (z. B. keine Kosten-Nutzen-Überlegungen). Es ist den Mitgliedern des AK Risikoableitung jedoch bewusst, dass bei der Auswahl von vielen Maßstäben (z. B. Adversitätsdefinition, zu Grunde gelegtes Vertrauensintervall, Einschluss oder Ausschluss von einzelnen Extrapolationsmodellen, Interpretation des Vorsorgebegriffs) implizit durch das Wissenschaftsverständnis Wertungen eingehen, die nicht alleine naturwissenschaftlich begründet sind.
1.2 Gültigkeit
(1) Die Regeln dieses Leitfadens beziehen sich ausschließlich auf eine Risikoquantifizierung für krebserzeugende Stoffe im Rahmen der Umsetzung der Gefahrstoffverordnung. Das unter Verwendung dieses Leitfadens quantifizierte Krebserkrankungsrisiko soll auch für die Ableitung eines Arbeitsplatzgrenzwerts (AGW) für krebserzeugende Stoffe nach § 3 Abs. 6 der GefStoffV herangezogen werden.
(2) Für den vorgesehenen Zweck sollen mit Hilfe dieses Leitfadens Expositions-Risiko-Beziehungen nach einheitlicher und transparenter Methodik geschätzt werden. Dabei geht es insbesondere um die Extrapolation von Risiken in den Niedrigrisikobereich bei limitierter Datenlage. An der Höhe des so ermittelten Risikos können sich Maßnahmen des Risikomanagements orientieren.
Somit wird die Möglichkeit eröffnet, dass das Ergebnis der Risikoquantifizierung nicht nur eine Punktschätzung des Risikos beinhaltet, sondern auch die Expositions-Risiko-Beziehung über einen weiten Bereich abbildet. Damit kann der Leitfaden auch bei Anwendung eines Drei-Bereiche-"Ampelmodells" (zwei Zäsurpunkte statt ein Grenzwert)(6)genutzt werden und die Expositions-Risiko-Beziehungen können die Aufstellung von "Verfahrens- und stoffspezifischen Kriterien" (VSK) nach § 9 Abs. 4 GefStoffV unterstützen (Bundesministerium für Arbeit und Soziales, 2005).
(3) Sonstige methodische Aspekte der Ableitung eines AGW für krebserzeugende Stoffe sowie die Ausweisung der Höhe des für einen AGW zu Grunde gelegten Krebserkrankungsrisikos sind nicht Gegenstand dieses Leitfadens, insbesondere nicht die Ausweisung der Höhe eines tolerablen und/oder akzeptablen Risikos.
Es ist somit nicht Gegenstand des Leitfadens, zu beantworten, bei welcher Risikohöhe der AGW liegen soll. Es soll jedoch möglich sein, in einem gesonderten Schritt regulatorisch relevante Zäsurpunkte in die ermittelte Expositions-Risiko-Beziehung einzufügen (z. B. Bedingungen für Ausnahmegenehmigungen, die an eine bestimmte Risikohöhe geknüpft sind).
Alle Risikoermittlungen beziehen sich im Übrigen auf das Auftreten einer Krebserkrankung sowohl bei tierexperimentellen Studien, bei denen (neben gestorbenen) auch die erkrankten Tiere erfasst werden, als auch bei Humandaten, für die ebenfalls Inzidenzdaten von Krebserkrankungen gegenüber Mortalitätsdaten bevorzugt werden. Fragen der Heilbarkeit von Tumorerkrankungen werden nicht berücksichtigt.
(4) Die Methodik dieses Leitfadens ist nicht 'dafür vorgesehen, tatsächliche Häufigkeiten von Krebserkrankungen für eine reale Arbeitsplatzsituation vorherzusagen oder entsprechende Hochrechnungen auf Erkrankungshäufigkeiten in der exponierten Bevölkerung vorzunehmen.
Es soll ausdrücklich vermieden werden, dass die Risikoquantifizierungen anderweitig missbräuchlich verwendet werden (z. B. um die Anzahl von expositionsbedingten Sterbefällen hochzurechnen). Die Expositions-Risiko-Modellierung, die Extrapolation auf niedrige Risiken und das unterstellte Expositionsszenario unterliegen bestimmten für eine harmonisierte Vorgehensweise in dem gegebenen Regulationsrahmen erforderlichen Konventionen, die jedoch nicht notwendigerweise für andere Zwecke adäquat sind. So muss dieser Ansatz z. B. für die Berechnung eines Kompensationsanspruchs nach der Berufskrankheitenverordnung nicht geeignet sein.
(5) Expositionsabschätzungen für einzelne Arbeitsplätze sind nicht Gegenstand dieses Leitfadens. Es wird nur ein Standardexpositionsszenario für den Arbeitsplatz unterstellt ("nominelles Risiko") (siehe Nummer 4.4).
1.3 Bedeutung der Standard-(Default-)annahmen
(1) Die Vorgaben in der Methodik dieses Leitfadens besitzen häufig Default-Charakter, d. h., sie sind dann heranzuziehen, wenn keine stoffspezifischen Informationen ein Abweichen vom Default rechtfertigen. Sollten jedoch stoffspezifisch solche qualifizierteren Daten vorliegen, kann begründet vom Default abgewichen werden. Die Begründung ist zu dokumentieren (siehe Nummer 8).
Erkenntnisse von geringer Relevanz reichen nicht immer aus, um ein Abweichen vom Default zu rechtfertigen. Zusätzliche Erkenntnisse können auch missbräuchlich für eine Risikoquantifizierung nach abweichender Methodik herangezogen werden: der hier offen gehaltene Ermessensspielraum ("kann abgewichen werden") erlaubt auch die Beibehaltung des Default und wird durch die geforderte Begründung eingegrenzt.
(2) In der Regel werden Schätzungen mit der relativ höchsten Wahrscheinlichkeit (zum Beispiel: geometrischer Mittelwert, "maximum likelihood"-Schätzung) zur Bildung des Default herangezogen.
Es wird ausdrücklich darauf verzichtet, bei allen Parametern "(reasonable) Worst case"-Annahmen vorzusehen. Bei der Auswahl handelt es sich um einen schwierigen Abwägungsprozess, der jedoch transparent zu gestalten ist. Das hier gewählte differenzierte Vorgehen wird vor dem Hintergrund der relativ hohen Unsicherheit bei den im Rahmen der Methodik vorzunehmenden Extrapolationsschritten gewählt, die derzeit mit keinem wissenschaftlichen Verfahren (z. B. einer Probabilitätsrechnung) vermindert werden kann. Bei Kombination zahlreicher "Worst case"-Annahmen würden zu einer Risikoquantifizierung mit sehr konservativem Charakter führen. Das Ergebnis lässt sich nicht validieren und verliert sich zunehmend im Spekulativen. Um die Begründungsdiskussion auf die eigentliche Risikoschätzung zu zentrieren statt auf die geeignete Bemessung des objektiv nicht näher eingrenzbaren Unsicherheitsbereichs, wird im vorliegenden Rahmen die angegebene Konvention gewählt.
(3) Die Bewertung der Daten zu Einzelstoffen und die sich daraus ergebenden Schlussfolgerungen (zum Beispiel zum anzunehmenden Wirkprinzip, Ausmaß der Abweichung vom Default-Wert im Einzelfall) ist nicht Gegenstand dieser Methodik.
Das stoffspezifische Vorgehen erfolgt - so weit es vom hier formulierten Default-Vorgehen abweicht - nach Maßstäben, die für den Einzelstoff zu begründen sind.
1.4 Definition und Einordnung der Risikozahl
(1) Dieser Leitfaden befasst sich mit den Methoden der Berechnung einer Risikozahl. Die Risikozahl stellt einen unter bestimmten Annahmen und für die einleitend definierten Zwecke berechneten Wert für das expositionsbedingte Lebenszeitrisiko im Szenario einer Exposition über das gesamte Arbeitsleben dar (definiertes Expositionsszenario siehe Nummer 4.4). Das Lebenszeitrisiko gibt die Wahrscheinlichkeit an, im Laufe des Lebens an einer bestimmten Tumor bzw. Krebsart zu erkranken, wenn die Sterblichkeit an anderen Ursachen ungefähr gleich ist wie in einer nicht-exponierten Population. Die Risikozahl kann auch als (statistischmathematische) Schätzung des Exzess-Risikos bzw. als "additional risk" oder "extra risk" bezeichnet werden, da dabei die Hintergrundinzidenz entsprechend eingerechnet wurde (siehe Nummer 3.5).
Die Aussagekraft des im Tierexperiment ermittelten Exzess-Risikos für ein Exzess-Risiko beim Menschen halten verschiedene Wissenschaftler für so gering, dass sie eine Risikoquantifizierung wegen zu großer Unsicherheiten auf dieser Basis ablehnen. Die Autoren dieses Leitfadens unterstützen mit einer Ausnahme jedoch die Verwendung der Risikozahl mit der Interpretation als Exzess-Risiko, wobei ausdrücklich auf die Definition (expliziter Ausweis der Randbedingungen des berechneten Risikos und der Unsicherheit) und die Abgrenzung gegenüber einem tatsächlich beim Menschen beobachtbaren Risiko verwiesen wird.
Der Begriff Lebenszeitrisiko soll deutlich machen, dass die gesamte Zeitspanne bis ins hohe Alter betrachtet wird, wobei eine Verteilung der Lebenszeiten wie in einer Allgemeinbevölkerung bzw. in der Kontrollgruppe eines Kanzerogenitätsversuchs zu Grunde gelegt wird (Becher und Steindorf, 1993). In der Praxis der quantitativen Risikoabschätzung bezieht sich die Ableitung des Risikos aber in der Regel auf ein ganz bestimmtes Alter, in Tierversuchen auf ungefähr 2 bis 2,5 Jahre, bei epidemiologischen Daten auf 70 bis 90 Jahre (z. B. 89 J.: Goldbohm et al, 2006; 85 J.: Attfield und Costello, 2004; Rice et al, 2001; SCOEL, 2003; Sorahan et al, 1998; Stayner et al, 1998, 2000; 80 J: HEI-AR, 1991; 75 J.: Stayner et al., 1995; Steenland et al, 2001). Das Statistische Jahrbuch 2006 für die Bundesrepublik Deutschland (Statistisches Bundesamt Deutschland, 2006) enthält durchschnittliche Lebenserwartungen, die anhand der altersspezifischen Mortalitätsraten der Jahre 2002/2004 berechnet wurden. Demnach reichte die statistische Lebenserwartung (ab dem Alter 20 Jahre) für Männer bis zum Alter von 76 Jahren und für Frauen bis zum Alter von 82 Jahren. Das Krebsrisiko nach der Sterbetafelmethode sollte daher mindestens bis zum Alter von 80 Jahren berechnet werden.
Das Risikomanagement kann sich, zusätzlich zu den Risikozahlen, auch auf das ALARA-Prinzip stützen ("as low as reasonably achievable"). Das ALARA-Prinzip alleine wird als unzureichend eingeschätzt, um Prioritäten im Umgang mit krebserzeugenden Stoffen differenziert zu erarbeiten. Grundsätzlich kann dem ALARA jedoch parallel gefolgt werden. Die Spezifizierung dieses Risikomanagement-Instruments ist nicht Gegenstand dieses Leitfadens.
(2) Statt durch Angabe eines "margin of exposure" (MoE; siehe Glossar, vgl. z. B. EC, 2006) wird im vorliegenden Konzept die in Abs. 1 definierte Risikozahl ausgewiesen; dies ermöglicht die Quantifizierung des nominellen Risikos für einen breiten Bereich der Expositions-Risiko-Beziehung.
Das Vorgehen, statt eines MoE eine Risikozahl (quantifiziertes Risiko) auszuweisen, resultiert auch aus dem Wunsch, für später zu berechnende AGW regelmäßig ein (angenommen) gleiches nominelles Risiko zu Grunde legen zu wollen (definiertes Schutzniveau). Für diese Einordnung ist es nicht ausreichend, einen MoE zu ermitteln.
In der Chemikalienbewertung mit MoE wird im abschließenden Schritt der Risikocharakterisierung
eine Quantifizierung vorgenommen (Abstand zwischen einer Prävalenz- z. B. als Benchmark-Dosis (10 Prozent)- und der Expositionshöhe wird berechnet),
dieser Abstand wird bewertet, also als " ausreichend" oder "nicht ausreichend" interpretiert. Bisher fehlen Regeln, wie sich eine über den "mode of action" angenommene Nichtlinearität in der Dosis-Risiko-Beziehung in der Interpretation dieses Abstandsmaßes niederschlagen sollte.
(3) Die Wahl der Risikozahl als Bewertungskriterium unterscheidet diesen Ansatz im Verständnis von dem Konzept der European Food Safety Authority (EFSA). Nach dem EFSA-Ansatz ergibt sich eine Punktschätzung (Angabe einer ausreichend sicheren Dosis oder Konzentration), während im vorliegenden Konzept die Expositions-Risiko-Beziehung über einen breiten möglichen Expositionsbereich definiert wird.
Während sich die Risikozahl am durchschnittlichen Risiko orientiert (empfindliche Personen sind geschützt, wenn das Risiko für durchschnittlich empfindliche Personen ausreichend gering ist), wird beim Konzept der EFSA versucht, den Schutz von empfindlichen Personengruppen durch Sicherheitsfaktoren explizit zu berücksichtigen. Bei ausreichender Höhe der Sicherheitsfaktoren wird - ähnlich dem Verständnis bei Annahme einer Wirkungsschwelle - kein noch verbleibendes Risiko quantifiziert (vgl. EFSA, 2005).
Bei den Leitfäden für die Erstellung eines "Stoffsicherheitsberichts" (CSR) im Rahmen der Chemikalienpolitik (REACH) wird bei der Ausweisung eines DM EL ("derived minimal effect level") entweder die Verwendung der Risikozahl vorgeschlagen (hier vorgesehenes Verfahren) oder alternativ die Herangehensweise nach EFSA (modifiziert) angewandt. Das EFSA-Verfahren ist ursprünglich für die Beschreibung eines erforderlichen Abstandes zwischen Prävalenz im experimentellen Szenario und Expositionshöhe nach oraler Aufnahme vorgesehen und nicht für den Arbeitsplatz bestimmt (andere Sicherheitsfaktoren), kann jedoch entsprechend angepasst werden. Für die Höhe der im modifizierten EFSA-Verfahren herangezogenen Sicherheitsfaktoren (Interspeziesvariabilität, Intraspeziesvariabilität, weitergehende individuelle Unterschiede in Krebsabwehrmechanismen) fehlen derzeit unterstützende statistische Daten oder Regeln. Verwendete Konventionen (ein Prozent Risiko für empfindliche Personen avisiert) wären gesellschaftlich zu konsentieren. Es wäre erforderlich, Maßstäbe zu erarbeiten, wie substanzspezifisch vom Default-Vorgehen abgewichen werden kann, wenn qualifiziertere Angaben vorliegen (Differenzierung im Vorgehen für verschiedene "mode of action"). Der nach dem modifizierten EFSA- Verfahren standardmäßig berechnete Grenzwert (DMEL) kann jedoch im Ergebnis mit einem DM EL identisch sein, der nach dem Konzept der Risikozahl berechnet wurde. Für die Anwendung der Risikozahl und die Transformation in einen DM EL im Rahmen von REACH fehlt derzeit die gesellschaftliche Konsentierung einer tolerablen und/oder akzeptablen (nominellen) Risikohöhe (die Ausweisung dieser Risikohöhe ist auch für die Anwendung des vorliegenden Leitfadens in nationaler Anwendung dann notwendig, wenn z. B. ein Arbeitsplatzgrenzwert für krebserzeugende Stoffe etabliert werden soll).
1.5 Datenbasis
(1) Sofern Humandaten zur Risikoquantifizierung vorliegen, so sind diese prioritär auf ihre Eignung zur Risikoquantifizierung hin zu überprüfen und ggf. heranzuziehen, jedoch ist die Datenqualität (Erkrankungsdaten, Expositionsverlauf) zu berücksichtigen. Risikoquantifizierungen auf tierexperimenteller Basis und auf humanepidemiologischer Basis sind vergleichend gegenüberzustellen (Plausibilitätskontrolle mit Humandaten).
Epidemiologische Studien können nur verwendet werden, wenn Effekte (Tumoren) beim Menschen aufgetreten sind. Eine negative Epidemiologie kann in der Regel nicht zur Plausibilitätskontrolle eines positiven tierexperimentellen Befundes herangezogen werden. Zur Einordnung der Relevanz von Humandaten im Vergleich zum Tierexperiment vgl. auch Goldbohm et al. (2006).
(2) Die Vorgehensweise dieses Leitfadens berücksichtigt, dass in der Mehrzahl der Fälle nur tierexperimentelle Daten als Basis der Risikoquantifizierung herangezogen werden können; entsprechend gelten die Festlegungen in diesem Leitfaden für tierexperimentelle Daten, jedoch werden Humandaten methodisch gleich behandelt, wo keine anders lautende Vorgehensweise beim jeweiligen Quantifizierungsschritt angegeben ist.
(3) Nicht positive epidemiologische Studienergebnisse stellen in der Regel keinen Nachweis der Abwesenheit eines möglichen Risikos dar. Sie sind diesbezüglich mit der gebotenen Zurückhaltung und unter Berücksichtigung ihrer Eignung für die gegebene Fragestellung (statistische Power, Höhe der Exposition, Qualität der Expositionseinstufung) zu interpretieren.
Literatur:
Ahlbom et al. (1990); Doll und Wald (1994)
1.6 Datenqualität
(1) Bei gewährleisteter Mindestqualität (siehe Nummer 7 dieses Leitfadens) können in der Regel Risikoquantifizierungen vorgenommen werden. Qualitätsmängel und die daraus resultierende Unsicherheit sind jedoch beim jeweiligen Schritt der Risikoquantifizierung zu dokumentieren.
Es können nicht immer Studien mit heute möglicher oder wünschenswerter Qualität als Grundlage für die Risikoquantifizierung vorausgesetzt werden. Der Übergang zwischen Qualitätsmängeln der Datenbasis und Unsicherheiten, die inhärent im Prozess der Risikoquantifizierung bei unvollständigem Wissen enthalten sind, ist fließend. Daher kann nur ein Abschneidekriterium definiert werden, wann die Gesamtunsicherheit (aus schlechter Datenlage plus Risikoquantifizierung mit Extrapolationsschritten) so groß ist, dass die resultierende Aussage als spekulativ und damit nicht mehr verwendbar zu bezeichnen ist (siehe Nummer 7). Der Umgang mit Unsicherheiten ist - darüber hinaus - bei dem jeweiligen Einzelschritt der Risikoquantifizierung und in Nummer 1.3 des Leitfadens festgelegt.
2 Diskussion des vorherrschenden Wirkprinzips
2.1 Wirkprinzip als Leitgröße zur Risikoquantifizierung
(1) Erkenntnisse zu dem vorherrschenden Wirkprinzip ("mode of action") bzw. den vorherrschenden Wirkprinzipien bei der beobachteten krebserzeugenden Wirkung einer Substanz sind sowohl für die Ermittlung des "point of departure" (Nummer 3) wie für die Durchführung der Extrapolation in den Bereich niedriger Risiken (Nummer 5) hilfreich. Entsprechend sind vor allem zu charakterisieren:
- 1.
die Art einer ggf. vorliegenden gentoxischen Wirkung,
- 2.
die Art nichtgentoxischer Ereignisse als Einflussgrößen auf den multifaktoriellen Prozess der Kanzerogenese,
- 3.
die jeweilige Bedeutung dieser Faktoren für das Wirkprinzip der Kanzerogenese und die Unsicherheit der entsprechenden Schlussfolgerung.
Die Ergebnisse sind in geeigneter Weise zu dokumentieren (Nummer 8).
2.2 Primäre und sekundäre Gentoxizität
(1) Es ist zu prüfen, ob eine unmittelbare Interaktion der Substanz mit dem Erbgut belegt oder aufgrund anderer Informationen anzunehmen ist. Sekundäre Gentoxizität (z. B. über oxidativen Stress, Interferenz mit dem mitotischen Prozess, Inhibition der Topoisomerase, Inhibition der DNA-Reparaturenzyme usw.) ist gegenüber der primären Gentoxizität (direkt, indirekt: DNA-Interaktion, Adduktbildung und Mutationen durch Muttersubstanz bzw. Metaboliten) zu unterscheiden.
(2) Die Qualität und die Absicherung der Einschätzung gentoxischer Eigenschaften ist zu charakterisieren (Differenzierung nach In-vivo-, In-vitro-Befunden, Kompatibilität der vorliegenden Studienergebnisse, Einfluss des Dosisbereichs in vorliegenden Tests, Information über Lücken).
(3) Informationen zur Gentoxizität (Art der Gentoxizität, Qualität und Absicherung der Erkenntnisse) können im Hinblick auf eine Spezifität am Zielorgan, in dem Tumorgenität beobachtet wurde, wesentlich sein. Bei manchen Formen der Gentoxizität (z. B. Aneuploidien) können Mindestschadstoffkonzentrationen angenommen werden, die erforderlich sind, um Krebs zu erzeugen.
Bei der Bewertung von Gentoxizitätstest ist zu bedenken, dass bis zu 80 Prozent der Stoffe, die negativ in Kanzerogenitätstests an Nagern sind, in einem oder mehreren In-vitro-Tests positiv sind. Dies betrifft vor allem Chromosomenaberrationstests, Mikrokerntests und den Maus-Lymphom-Test. Hierfür gibt es in Abhängigkeit vom verwendeten Testsystem und von der Substanzklasse zahlreiche Gründe, die die Übertragbarkeit der In-vitro-Ergebnisse auf die In-vivo-Situation nicht gestatten und von denen einige beispielhaft aufgeführt werden:
Verwendung hoher Konzentrationen, die metabolische Detoxifizierungsmechanismen überlasten,
im Testsystem fehlende Phase-II-Enzyme und deren Kofaktoren,
Testsystem mit DNA-Reparatur-Defizienz (alle Salmonella-Stämme und E. coli),
Testsystem ohne oder mit anomaler Expression von p53-Protein (CHO-Zellen, L5178Y-Zellen, V79-Zellen),
Effekte mit Schwelle, die in vivo nicht erreicht wird: An-euploidie, Hemmung der DNA-Polymerase, der Topoisomerasen oder Kinasen, Zytotoxizität, pH-Wert-Änderung.
Weiterhin ist die Übertragbarkeit auf den Menschen eingeschränkt durch Verwendung rattenspezifischer metabolischer Aktivierung, die nicht das Muster fremdstoffmetabolisierender aktivierender Enzyme beim Menschen widerspiegelt (Kirkland et al, 2007a). Andererseits ist es auch möglich, dass die Aktivierung im Organismus nicht in Standard-in-vitro-Tests nachgebildet wird, z. B. bei Aktivierung des Stoffs über Sulfotransferasen und daher falsch-negative Ergebnisse erhalten werden (Kirkland et al., 2007b).
Um zu entscheiden, ob ein kanzerogener Stoff primär gentoxisch wirkt, ist daher die Relevanz der Ergebnisse von In-vitro-Gentoxizitätstests anhand der in den Tests verwendeten Bedingungen (z. B. Vergleich der Dosis-Wirkungs-Beziehungen von Gentoxizität und Zytotoxizität, Hochdosiseffekte) und der Struktur des untersuchten Stoffs zu prüfen. Gegebenenfalls sollten Struktur-Wirkungs-Beziehungen miteinbezogen werden. Bei systemisch wirkenden Kanzerogenen geben im Zweifelsfall die Ergebnisse von validen In-vivo-Tests den Ausschlag. Bei lokal wirkenden Kanzerogenen sind negative In-vivo-Tests nur dann aussagekräftig, wenn gezeigt wird, dass das Zielorgan erreicht wurde.
2.3 Nichtgentoxische Ereignisse
(1) Informationen zu nichtgentoxischen Effekten mit möglicherweise ursächlichem Einfluss auf den Prozess der Kanzerogenese sind zu erfassen und zu beschreiben sowie der ermittelte Dosisbereich mit dem krebsauslösenden Dosierungen zu vergleichen. Zu nennen sind insbesondere Zytotoxizität (z. B. Reizung, Entzündung, Nekrose), induzierte Zeilproliferation, toxikokinetische Informationen (z. B. Enzyminduktion, Sättigung bzw. neue Metaboliten spezifisch bei hoher Dosis), rezeptorvermittelte Prozesse, Proteinbindung, direkte hormonelle Wirkung, indirekter Einfluss auf Hormonregelkreise, Organspezifität und Geschlechtsspezifität.
(2) Die Qualität und die Absicherung der Einschätzung nichtgentoxischer Eigenschaften ist zu charakterisieren (Differenzierung nach In-vivo-, In-vitro-Befunden, Kompatibilität der vorliegenden Studienergebnisse, Einfluss des Dosisbereichs in vorliegenden Tests, Information über Lücken).
(3) Informationen zu nichtgentoxischen Ereignissen (Art des Effekts, Qualität und Absicherung der Erkenntnisse) sind insbesondere in Bezug auf die Relevanz im Zielorgan, in dem Tumorgenität beobachtet wurde, zu spezifizieren.
2.4 Bedeutung verschiedener Einflüsse im multifaktoriellen Geschehen
(1) Nach einem "Weight of evidence"-Ansatz sind die Bedeutung der primären und/oder sekundären Gentoxität (siehe Nummer 2.2) und nichtgentoxischer Ereignisse (siehe Nummer 2.3) auf den Prozess der Kanzerogenese abzuschätzen. Der oder die zentralen Einflussfaktoren auf das Krebsgeschehen sind darzustellen und deren vermutete Bedeutung für den Menschen zu begründen.
(2) Ergebnis kann auch eine, je nach Tumorlokalisation und/ oder Dosisbereich differenzierte, Unterscheidung der anzunehmenden Wirkprinzipien sein. Das Vorliegen mehrerer (möglicher) Wirkprinzipien ist kenntlich zu machen.
(3) Das Vorliegen prämaligner Effekte (wie die Bildung von Foci in der Leber) ist zu prüfen und deren Dosis-Wirkungsbeziehung nach Möglichkeit zu beschreiben.
(4) Hintergrundraten und das Auftreten spontaner Tumoren in der Kontrollgruppe sind bei der Diskussion des Wirkprinzips einzuordnen.
2.5 Zielgerichtete Schlussfolgerung
(1) Die Erfassung des Informationsstands mündet in folgenden Aussagen:
postuliertes Wirkprinzip
Schlüsselereignisse (beobachtete, Übereinstimmung mit Wirkprinzip)
Dosis-Wirkungs-Zusammenhang
zeitliche Assoziation
Stärke des Zusammenhangs, Konsistenz der Daten für diese Schlussfolgerung, Spezifität der Assoziation
biologische Plausibilität
andere mögliche Wirkprinzipien
Vertrauen in die Einschätzung
Datenlücken, Unsicherheiten.
(2) Es ist insbesondere zu beantworten:
Ist das "weight of evidence" ausreichend, um im Tierexperiment ein Wirkprinzip zu benennen?
Kann die Humanrelevanz des Wirkprinzips mit hinreichender Wahrscheinlichkeit ausgeschlossen werden auf Basis grundsätzlicher qualitativer Unterschiede in Schlüsselereignissen zwischen Tier und Mensch?
Kann die Humanrelevanz des Wirkprinzips mit hinreichender Wahrscheinlichkeit auf Basis quantitativer toxikokinetischer und/oder toxikodynamischer Unterschiede zwischen Tier und Mensch ausgeschlossen werden?
Welches Vertrauen besteht in die abgegebene Einschätzung (Bedeutung)?
Auch bei gentoxischen Ereignissen kann eine Wirkungsschwelle auftreten. Gentoxische Ereignisse sind unter diesem Blickwinkel zu differenzieren (vgl. TGD, Risk Characterisation, Abschnitt 4.14.3.4; Butterworth, 2006).
Auch nicht gentoxische Ereignisse können nicht regelmäßig mit einer Wirkungsschwelle verknüpft werden, z. B. ist bei einigen Rezeptor-vermittelten Prozessen der Ausweis eines Werts für eine solche Wirkungsschwelle nicht immer möglich (vgl. TGD, Risk Characterisation, Abschnitt 4.14.3.3; Butterworth, 2006).
Soweit für die Ermittlung der Bedeutung der verschiedenen Aussagen Angaben zur Expositions-Risiko-Beziehung im experimentellen Bereich erforderlich sind, besteht eine Interdependenz zwischen Aufgaben nach Nummer 3 und Aufgaben nach Nummer 2 dieses Leitfadens (insbesondere 2.4 und 2.5: Expositions-Risiko-Beziehung). Entsprechend können die Positionen dieses Leitfadens nicht in strenger zeitlicher Abfolge bearbeitet werden.
Die angesprochenen Punkte unter 2.5 basieren auf Überlegungen der WHO (IPCS) und sind im Detail in Boobis et al. (2006) erläutert. Beispiele für die Vorgehensweise bei der Diskussion des Wirkprinzips finden sich z. B. in Kirman et al. (2004), Cohen et al. (2003) sowie Preston und Williams (2005). Die grundsätzliche methodische Vorgehensweise, um den "mode of action" zu erfassen, ist in Meek et al. (2003) und Seed et al. (2005) erläutert.
In verschiedenen Veröffentlichungen (z. B. Streffer et al., 2004; Hengstler et al., 2006; Bolt und Huici-Montagud, 2007; Foth et al, 2005) wurden ähnliche Differenzierungen des Wirkprinzips gefordert, wie sie sich in der hier zu Grunde gelegten Vorgehensweise darstellen. Diese münden in einer Differenzierung wie sie auch in Nummer 5.1 dieses Leitfadens vorgenommen wurde.
Neumann (2006a, b, c) begründet die Unmöglichkeit, bei krebserzeugender Wirkung eine eindeutige Schwelle zu finden und schlägt vor, den Begriff gänzlich zu vemeiden. Wegen der nicht vorliegenden besser kommunizierbaren Alternativen wird jedoch im vorliegenden Rahmen mit den oben ausgeführten Einschränkungen im Verständnis der Begriff weitergeführt.
3 Risikoquantifizierung im Bereich beobachteter Krebsinzidenzen
3.1 Auswahl von Tierspezies, Geschlecht und Tumorlokalisation(en)
(1) Liegen Tumordaten zu mehreren der üblicherweise eingesetzten Tierarten vor, so sind diejenigen zu der Tierspezies bevorzugt heranzuziehen, die am empfindlichsten reagiert.
(2) Bei der Auswahl der Tierspezies und der dort beobachteten Tumortypen und -lokalisationen ist jedoch abzuwägen, inwieweit eine quantitative Übertragbarkeit auf den Menschen angenommen werden kann. Eine Übertragbarkeit ist insbesondere dann anzunehmen, wenn die Tumorlokalisation im Speziesvergleich identisch ist und/oder Erkenntnisse zum "mode of action" das Auftreten eines bestimmten Tumortyps (oder einer bestimmten Tumorlokalisation) stützen.
Tierexperimentelle Studien werden vor dem Hintergrund durchgeführt, dass qualitative und quantitative Übertragungen auf dem Menschen (ggf. unter Berücksichtigung von Extrapolations- oder Korrekturfaktoren) prinzipiell möglich sind. Insofern ist grundsätzlich das tierexperimentelle Modell mit der größten Verwandtschaft zum Menschen zu bevorzugen. Im Falle des Nichtwissens darüber, welches Tiermodell im speziellen Fall dem Menschen am nächsten steht, ist ein konservatives Herangehen der geeignete Maßstab. Dieses gilt grundsätzlich, auch wenn im Einzelfall Widersprüche aufgezeigt wurden: Bei 1,3-Butadien scheint der menschliche Metabolismus dem der weniger empfindlichen Ratte ähnlicher zu sein als dem der empfindlicheren Maus. Werden jedoch epidemiologische und tierexperimentelle Risikoquantifizierungen gegenübergestellt, ist bei 1,3-Butadien eine größere Übereinstimmung des Krebsrisikos für Maus und Mensch zu beobachten (Roller et al, 2006). Dieser mögliche Widerspruch im Falle von 1,3-Butadien bedeutet, dass
- 1.
den Humandaten besonderes Gewicht zuzumessen ist (siehe Nummer 1.5 Abs. 1),
- 2.
konservative Extrapolationsschritte wie die Annahme von Linearität im Niedrigrisikobereich nicht vorschnell wegen vermeintlicher mechanistischer Hinweise aufgegeben werden sollten, und
- 3.
die relative Empfindlichkeit von Versuchstieren gegenüber dem Menschen weiterer Überprüfung bedarf.
(3) Eine im Tierexperiment beobachtete Tumorlokalisation, die von den Beobachtungen aus epidemiologischen Studien beim Menschen abweicht, spricht in der Regel nicht gegen deren Humanrelevanz (siehe aber Hinweis unter Nummer 3.1 Abs. 6). Die resultierende Risikoquantifizierung ist jedoch als unsicherer zu betrachten.
(4) Liegen erhöhte Tumorinzidenzen in beiden Geschlechtern vor, so sind in der Regel die Daten zu jenem Geschlecht mit der höheren Tumorrate heranzuziehen. Liegen die Tumorraten in beiden Geschlechtern etwa in gleicher Höhe, so ist zur Erhöhung der statistischen Absicherung eine Addition der Daten zu beiden Geschlechtern zulässig.
(5) Liegen Tumore in mehreren Organen vor, so sind die Daten zu allen den Organen heranzuziehen, bei denen eine statistisch und/oder biologisch erhöhte Tumorenzahl in einer Dosierung beobachtet wird, und/oder eine statistisch signifikante Dosis-Wirkungsbeziehung (ggf. auch nur als Trend) erkennbar ist.
Es gibt eine Reihe typischer Tumorformen, die in bestimmten Nagerstämmen mit hoher, teilweise auch stark variabler Spontaninzidenz auftreten und deren Relevanz für den Menschen nicht feststeht (siehe Nummer 3.1 Abs. 6). Wenn deren Häufigkeit dosisabhängig gegenüber der aktuellen und der mittleren historischen Kontrolle erhöht ist, wird man in der Regel von einem expositionsbedingten Effekt sprechen.
Zunächst ist zu prüfen, ob nicht auch andere Tumorformen aufgetreten sind, die keinesfalls der Spontanpathologie zugeordnet werden können und ob diese nicht bei noch niedrigerer Dosis und/oder in größerer Häufigkeit aufgetreten sind und allein schon aus diesem Grunde als Berechnungsgrundlage vorgezogen werden sollten.
(6) Die Berücksichtigung oder Nichtberücksichtigung bestimmter Tumorlokalisationen (ggf. mit Einschränkung auf bestimmte Tierspezies oder -stämme) stellt eine Einzelfallabwägung dar. Als Hinweise für die Frage der (qualitativen und/oder quantitativen) Übertragbarkeit auf den Menschen gelten:
Es ist keine (qualitative oder quantitative) Übertragbarkeit anzunehmen: alpha-2u-globulinbedingte Nierentumoren der männlichen Ratte.
In der Regel nur qualitativ (nicht quantitativ) übertragbar, wenn auch zugleich Gentoxizität vorliegt (in der Regel auch nicht qualitativ übertragbar, wenn keine Gentoxizität vorliegt): Lebertumoren nach PPAR(alpha)-Rezeptor-Stimulation ("Peroxisomenproliferation"), Leukämien bei Fischer-344-Ratte, Phäochromocytome, wenn diese nur bei männlicher F344-Ratte auftreten, Vormagentumoren und Tumoren der Zymbal-, Harderschen Drüse sowie Klitoris- und Präputialdrüse, wenn außer an diesen Lokalisationen keine Tumoren in anderen Lokalisationen auftreten.
Der rein qualitative Speziesvergleich ist für Einstufungen relevant, jedoch nicht für die hier betrachtete Ermittlung einer Expositions-Risiko-Beziehung und einer Konzentration in Bezug auf eine bestimmte Risikozahl.
In der Regel quantitativ übertragbar, wenn zugleich Gentoxizität vorliegt, jedoch mit erhöhter Unsicherheit (d. h. in der Regel nur qualitativ übertragbar, wenn keine Gentoxizität vorliegt): Leydigzelltumoren bei Nagern, Lebertumoren bei B6C3F1-Maus, Phäochromocytome bei F344-Ratte, wenn diese in beiden Geschlechtern auftreten (Differenzialdiagnostik zu - altersbedingten -Hyperplasien beachten, Daten zu weiblichem Tier für Quantifizierung geeigneter), Schilddrüsentumoren bei der Ratte, Vormagentumoren und Tumoren der Zymbal-, Harderschen Drüse sowie Klitoris- und Präputialdrüse, wenn außer an diesen Tumorlokalisationen zugleich Tumoren in anderer Lokalisation auftreten.
Auch ohne Gentoxizität quantitativ in der Regel übertragbar, teilweise jedoch mit erheblichen Unsicherheiten: alle anderen Lokalisationen und Tumorarten, Tumoren bei anderen als den genannten Tierspezies oder -stämmen.
Bei der Abwägung, ob eine quantitative Übertragbarkeit angenommen wird, ist die (beobachtete oder zu unterstellende) Konzentration der Substanz am Zielorgan einzubeziehen.
Bei der Abwägung, ob eine quantitative Übertragbarkeit angenommen wird, ist das zu unterstellende Wirkprofil (siehe Nummer 2) einzubeziehen.
Liegen sowohl Tumorinzidenzen an
- 1.
Lokalisationen mit fraglicher Humanrelevanz und/ oder fraglicher quantitativer Übertragbarkeit vor und an
- 2.
Lokalisationen mit eindeutigerer quantitativer Übertragbarkeit, so ist letzteren in der Regel der Vorzug bei der Risikoquantifizierung zu geben.
Eine ausführlichere Diskussion zum Hintergrund dieser Differenzierung befindet sich in Anhang 10.3 dieses Leitfadens (mit Literaturhinweisen).
(7) Die Tumorinzidenzen in den verschiedenen Organen, die unter Abs. 5 und Abs. 6 ausgewählt wurden, sind in der Regel getrennt zu quantifizieren und vergleichend gegenüberzustellen. Die Risikoquantifizierung erfolgt im Standardfall mit derjenigen Tumorlokalisation mit der niedrigsten T25, d. h. eine Dosis oder Konzentration, bei der bei 25 Prozent der Tiere Krebs auftritt. Dabei wird die unterschiedliche Hintergrundrate bei der T25-Berechnung berücksichtigt. In Einzelfällen ist es jedoch geboten, auch verschiedene Tumorlokalisationen zusammenzufassen (Beispiel: Asbest - Mesotheliome, Lungentumoren). Im Falle, dass eine solche Zusammenfassung vorgenommen wird, ist die Maßgeblichkeit der Gesamtinzidenz für die Risikoquantifizierung zu begründen.
Bei T25-Verfahren wird ausgehend von einer Konzentration mit signifikant erhöhter Tumorinzidenz durch lineare Interpolation
- 1.
unter Berücksichtigung der Hintergrundinzidenz,
- 2.
gegebenenfalls unter Korrektur einer nicht lebenslangen Versuchsdauer, und
- 3.
unter Annahme einer vollständigen Resorption eine Dosis ermittelt, bei der die Inzidenz für diesen Tumor im Tierversuch 25 Prozent bei lebenslanger Exposition beträgt (vgl. auch Glossar).
Mit der Berechnung von T25 oder BMD für mehrere Tumorlokalisationen, Geschlechter sowie mit und ohne gutartige Tumoren soll ermöglicht werden, in späteren Schritten parallel von mehreren "points of departure" (siehe Nummer 3.2) aus und verknüpft mit einer differenzierten mechanistischen Diskussion in den Niedrigrisikobereich zu extrapolieren. Aggregationen (Zusammenfassungen von Befunden) sind insbesondere dann sinnvoll, wenn die Frage der Differenzierung verschiedener Dosis-Wirkungsbeziehungen (z. B. wegen der Homogenität der beobachteten Reaktionen) nicht im Vordergrund steht. So kann es sinnvoll sein, die Befunde bei einer einheitlichen Wirkungsweise eines Kanzerogens in verschiedenen Organen auch über verschiedene Tumorlokalisationen zu aggregieren. Im TGD der EU wird ausgeführt: "For a substance inducing more than one type of tumours, the determination of a dose-descriptor value is from each relevant tumour type rather than from the number of tumour bearing animals. If several relevant data sets on tumour-incidences are available, dose descriptors values should be derived for all these." (EC 2006, Abschnitt 4.14.2.3). Verschiedene Hintergrundraten von Tumoren in verschiedenen Organen sprechen gegen eine Aggregation mehrerer Tumorlokalisationen.
Für eine differenziertere Betrachtung der Möglichkeiten zur Zusammenordnung von Tumoren für die Krebsrisikoberechnung argumentieren McConnell et al. (1986). EPA interpretiert diese Auswertung: " The incidence of benign and malignant lesions of the same cell type, usually within a single tissue or organ, are considered separately and are combined when scientifically defensible" (Eine konkrete Auflistung, wann Zusammenordnungen vorgenommen werden können, wird in McConnell et al. gegeben).
Es wird also nicht das Prinzip vertreten, die Gesamtzahl der tumortragenden Tiere, gleich welcher Tumorlokalisation, aufzuaddieren.
Manche ältere Studien waren auch so angelegt, dass nur verdächtige Zielorgane ausgewertet wurden. Entsprechend selektive Studien können dennoch für die Risikoquantifizierung herangezogen werden, wenn sie kanzerogene Wirkungen erkennen lassen. Mehrfach-Tumoren (Multiplizität) werden üblicherweise zusätzlich berichtet, wenn sie beobachtet werden.
(8) Liegen in einem Organ/Gewebe mehrere Tumortypen vor, so ist in der Regel eine gemeinsame Betrachtung zu wählen. In begründeten Einzelfällen (z. B. Humanrelevanz nur eines Tumortyps) ist jedoch eine getrennte Betrachtung angezeigt.
(9) Liegen in einem Organ gutartige und bösartige Tumoren vor, so wird deren Inzidenz in der Regel addiert. Eine Addition verschiedener Tumortypen in einem Tier erfolgt jedoch nicht, da sonst eine Überschreitung der Gesamtinzidenz (bezogen auf das Organ > 100 Prozent) eintreten kann. Liegen Hinweise darauf vor, dass z. B. eine Malignisierung eines gutartigen Tumors beim Menschen unwahrscheinlich ist, kann begründet auf eine entsprechende Addition verzichtet werden.
3.2 Auswahl eines "point of departure"
(1) Der "point of departure" (POD; ein Ausgangspunkt für weitere Schritte der Risikoabschätzung) ist eine definierte Expositionshöhe mit Risikozuordnung auf der Konzentrations-Risiko-Funktion für eine Substanz. Der POD liegt auf oder nahe bei der Expositionshöhe (Konzentrationsbereich), zu der aus epidemiologischen oder aus tierexperimentellen Beobachtungen Daten über das Auftreten von Krebshäufigkeiten vorliegen. Für den POD wird das Risiko als Krebsinzidenz in Prozent der zugehörigen Konzentration (mg/m3) gegenübergestellt. Der POD ist ein normalisierter Wert. Unter "Normalisierung" ist die Umrechnung auf Lebens(arbeits-)zeitexposition (siehe Nummer 4.3), die Pfad-zu-Pfad-Extrapolation auf den Inhalationspfad (siehe Nummer 4.2) und die Berücksichtigung der Hintergrundinzidenz (siehe Nummer 3.4) in der vorgegebenen Weise zu verstehen. Der POD dient als Startpunkt für eine Extrapolation oder zu Vergleichszwecken; somit ist der T25 je nach Vergleichsebene bereits als Humanäquivalent anzugeben (hT25) oder auf der Ebene des Tierexperiments zu nutzen. Die Randbedingungen der Anwendung eines T25 sind jeweils präzise auszuweisen.
(2) Bei hinreichender Qualität der Beobachtungsdaten ist der POD als "Benchmark-Konzentration" bzw. Benchmark-Dosis auszuweisen. Dabei ist der zentrale Schätzwert (BMD) und nicht der 95-Prozent-Vertrauensbereich (BMDL) (7)heranzuziehen. (8) Der POD dient als Startpunkt für eine Extrapolation oder zu Vergleichszwecken; somit ist die Benchmark-Dosis je nach Vergleichsebene bereits als Humanäquivalent anzugeben (HBMD, HBMDL) (9) oder auf der Ebene des Tierexperiments zu nutzen. Die Randbedingungen der Anwendung einer Benchmark-Dosis sind jeweils präzise auszuweisen.
Die Kriterien für eine ausreichende Qualität der Daten zur Modellierung nach dem Benchmark-Verfahren sind gesondert festzulegen (siehe Nummer 3.3). Der Faktor zwischen BMD und BMDL gibt auch eine Aussage zur Qualität der vorgenommenen Modellierung (Anpassungsgüte der Modellfunktion an die vorliegenden experimentellen Daten). Insofern kann bei Berechnung der BMDL dieser Faktor auch (neben anderen Kriterien) für die Beurteilung der Frage herangezogen werden, ob das Benchmark-Verfahren im konkreten Fall überhaupt zur Anwendung kommen sollte.
Die Auswahl des BMD statt des BMDL beinhaltet möglicherweise einen gewissen Fehler (da nicht ausgeschlossen werden kann, dass die Expositions-Risiko-Beziehung durch den BMDL korrekter beschrieben wird). Die Wahl des BMD erscheint jedoch begründet
- 1.
wegen der Analogie zum T25 bei schlechterer Datenlage (T25 ist ebenfalls ein zentraler Schätzwert ohne Vertrauensbereich),
- 2.
wegen des nur geringen möglichen Fehlers (bei großer Abweichung zwischen BMD und BMDL würde dies gegen die Verwendung des Benchmark-Verfahrens sprechen),
- 3.
da durch die Linearisierung im Bereich unterhalb der BMD als POD in den meisten Fällen ohnehin ein konservatives Extrapolationsverfahren gewählt wird.
Zur Umrechnung einer Benchmark-Dosis auf eine äquivalente Humanexposition siehe Nummer 4.
(3) Die "benchmark response" beim POD ist aus Gründen der Vergleichbarkeit auf 10 Prozent zu setzen.
In vielen Fällen gibt es keine starken Abweichungen im angenommenen Risiko, wenn der T25 mit der BMDw unter Korrektur (lineare Umrechnung) des Risikoniveaus verglichen wird (vgl. Anhang zu EC, Technical Guidance Document, 2006). Je nach Verlauf der Konzentrations-Risiko-Beziehung sind jedoch Abweichungen möglich. Deshalb und wegen der kompletteren Beschreibung des abgeleiteten Verlaufs der Konzentrations-Risiko-Beziehung im experimentellen Bereich wird der Anwendung des Benchmark-Verfahrens der Vorzug gegeben. Zu Beispielen siehe Nummer 5.2.
Eine Fortführung der Modellierung zwischen BMD10 und BMD0,1 wird im vorliegenden Leitfaden für den Fall einer mechanistisch begründeten Nichtlinearität bei guter Datenlage eingesetzt (siehe Nummer 5.2). Liegen keine hinreichenden Gründe für Nichtlinearität vor, so wird die Modellierung mit Benchmark-Methode nur für den experimentellen Bereich bis zu einer BMD10 als POD vorgenommen. In der früheren Vorgehensweise der U.S. EPA wurde das linearisierte Multistage-(LMS-)Modell herangezogen. Dieses Verfahren ist praktisch identisch mit einer Modellierung mit dem Multistage-Modell im experimentellen Bereich und der Fortführung der modellierten Funktion in den Niedrigrisikobereich (z.B. bei "benchmark response" 1:1.000). Dabei wird jedoch der 95-Prozent-Vertrauensbereich einbezogen.
(4) Ist die Ausweisung einer hinreichend qualifizierten Benchmark-Konzentration nicht möglich, ist die T25 in der Berechnung nach dem Verfahren von Sanner et al. (2001)/ Dybing et al. (1997) als POD heranzuziehen.
Der T25 wird gegenüber ähnlichen anderen Werten als POD der Vorzug gegeben, wenn das Benchmark-Verfahren nicht eingesetzt werden kann, weil
dies dem Verfahren der Risikoquantifizierung in verschiedenen Festlegungen zum Risk Assessment der EU entspricht,
die in Deutschland früher diskutierte "Steinhoff"-Methode mit dem T25 als POD kompatibel ist, sie jedoch nicht auf einen normierten Prozentsatz (25 Prozent) bezogen ist,
die LED10 in den USA (EPA, 2005) die Anwendung des Benchmark-Verfahrens voraussetzt, was nicht immer hinreichend qualifiziert ist.
Das ED10-Verfahren der U.S. EPA basiert ebenfalls auf der Benchmark-Modellierung (ohne Berücksichtigung des Vertrauensbereichs) und ist methodisch identisch zur Ableitung der BMDw. Für die Berechnung eines Referenz-MoE (siehe Glossar "margin of exposure") nach EU/TGD wird in der Regel der Unterschied zwischen T25 und ED10 linear berücksichtigt, so dass in dem EU-MoE-Ansatz auch die ED10 als POD herangezogen werden kann.
(5) Für Extrapolationen in den Bereich unterhalb der beobachteten Inzidenzen, bei denen die Fortsetzung der Konzentrations-Wirkungs-Beziehung angenommen wird, wie diese im Beobachtungsbereich bereits vorliegt (stetige Funktion; siehe Nummer 5.2), ist die Angabe eines POD formal nicht erforderlich. Dieser sollte aber dennoch zu Vergleichszwecken ausgewiesen werden.
(6) BMD10 bzw. T25 sind für alle humanrelevanten Tumorlokalisationen zu errechnen (zur Auswahl der Tumorlokalisationen und Spezies siehe Nummer 3.1).
(7) Bei Benchmark-Modellierungen mit schlechterer Datenqualität (siehe Nummer 3.3) ist es sinnvoll, sowohl die Berechnung des T25 wie der BMD10 vorzunehmen, um die Auswirkungen der Unsicherheit der jeweiligen Entscheidung zu erkennen: ggf. liegen die nach den jeweiligen Verfahren ermittelten POD nahe beieinander oder zeigen deutliche Diskrepanzen. Die entsprechende Information ist zu dokumentieren.
Beispiele: siehe Nummer 5.2 (Fall B)
3.3 Mindestkriterien an Datenqualität für Anwendung des Benchmark-Verfahrens
(1) Zur Wahl des Benchmark-Verfahrens sollten in der Regel mindestens die Daten zur Kontrollgruppe und drei Dosisgruppen vorliegen.
In Annex XI zum TGD der EU wird anhand einiger Beispiele eine Abwägung zwischen T25 und BMD05 vorgenommen. Dabei wurde das genannte Kriterium hervorgehoben.
(2) Unterscheidet sich die Tumorhäufigkeit in allen Dosisgruppen nicht oder nur unwesentlich (Plateaueffekt), ist die Anwendung des Benchmark-Verfahrens nicht sinnvoll.
(3) Gibt es nur eine Dosisgruppe außer der Kontrolle, bei der die Effektstärke deutlich über der BMR (10) liegt, ist das Benchmark-Verfahren nicht sinnvoll anwendbar.
(4) Liegt die Tumorhäufigkeit in nur einer Dosisgruppe unter 100 Prozent Inzidenz (außer bei der Kontrolle), ist das Benchmark-Verfahren nicht sinnvoll anwendbar.
(5) Das Benchmark-Verfahren ist nicht anwendbar, wenn die Modellierung mit den vorliegenden Daten eine zu schlechte Anpassung erlaubt (Modellfit: p < 0,1, Chi-Quadrat außerhalb - 2 bis + 2). Ferner ist die Unsicherheit der Abschätzung zu groß, wenn beim betrachteten BMR das Verhältnis BMD/BMDL > 10 ist.
Im Abschlussbericht des Projekts FKZ 201 65 201/01 ("Vergleich der Verfahren zur Ableitung gesundheitsbezogener Wirkungsschwellen" Benchmark - NOAEL, Umweltbundesamt 2003) werden die weiteren Kriterien gemäß Absatz 2-5 diskutiert und begründet.
(6) Für Zweifelsfälle mit begrenzter Datenqualität ist das Vorgehen nach Nummer 3.2 Abs. 7 zu wählen, nämlich zwischen T25 und dem Benchmark-Verfahren abzuwägen. Die Begründung für die Verfahrensweise ist zu dokumentieren.
Beispiel: siehe Nummer 5.2 (Fall B)
3.4 Anwendung des Benchmark-Verfahrens
(1) Die für die Kurvenanpassung auszuwählenden Modelle sollten den mechanistischen Vorstellungen zur Kanzerogenese nicht widersprechen. Deshalb wird oft das Multistage-Modell herangezogen, das dem Mehrstufenmodell der Krebsentstehung entspricht. Die Gamma-Funktion entspricht jedoch ebenfalls dem mechanistischen Verständnis eines "Multi-Hit"-Modells zur chemischen Kanzerogenese. Multistage- oder Gamma-Funktion sind demnach die bevorzugten Modelle zur Modellierung mit dem Benchmark-Verfahren im experimentellen Bereich. Andere Modelle sollten jedoch nicht ausgeschlossen werden, wenn sie eine deutlich bessere Datenanpassung ermöglichen. Modelle mit einer ähnlichen Anpassungsgüte, die jedoch weniger Parameter für die Modellierung benötigen, sind zu bevorzugen (erkennbar am AlC-Wert in der Ergebnisdarstellung der entsprechenden Software der U.S. EPA). Die Qualität der Datenpassung ist wichtiger im Bereich niedriger experimenteller Konzentrationen als im Bereich hoher Konzentrationen.
Im Abschlussbericht des Projekts FKZ 201 65 201/01 ("Vergleich der Verfahren zur Ableitung gesundheitsbezogener Wirkungsschwellen" Benchmark - NOAEL, Umweltbundesamt 2003) werden die genannten Kriterien diskutiert und begründet.
3.5 Umgang mit Hintergrundinzidenz
(1) Entsprechend dem Standardvorgehen beim T25-Ansatz und beim Benchmark-Verfahren (nach Software der U.S. EPA) ist in der Regel der "Extra risk"-Ansatz heranzuziehen.
Die Konvention, das "extra risk" zu wählen, ist aus toxikologischer Sicht nicht gut begründet, wird jedoch als Standardvorgehen akzeptiert, da
- 1.
in der Regel die Abweichungen bei niedriger Hintergrundrate gering ausfallen,
- 2.
eine Übereinstimmung mit vielen älteren unit risk-Berechnungen entsteht,
- 3.
eine Übereinstimmung mit dem T25-Ansatz so gewährleistet ist,
- 4.
eine Übereinstimmung mit der traditionellen Vorgehensweise beim Multistage-Verfahren so gewährleistet ist.
(2) Bei sehr hohen Inzidenzen in der Kontrollgruppe oder beim Vergleich mit Humandaten ist das "additional risk" heranzuziehen und dieses Vorgehen zu begründen.
3.6 Risikoquantifizierung durch Ausweisung des T25
(1) Die Festlegung eines POD durch die Ausweisung des T25-Wertes nach dem Verfahren von Sanner et al. (2001) und Dybing et al. (1997) erfordert keine Modellierung der Dosis-Wirkungsbeziehung im experimentellen Bereich. T25 wird durch lineare Interpolation bestimmt. Dieses Verfahren ist regelmäßig heranzuziehen, wenn eine qualifizierte Benchmark-Berechnung nicht möglich ist.
Zur näheren Definition des T25 vgl. Glossar.
(2) Wenn ausschließlich der Inhalationspfad relevant ist (für Arbeitsplatzgrenzwerte der Fall), wird der T25-Wert als Luftkonzentration (mg/m bzw. ppm) ausgedrückt.
Zur weiteren Normierung des T25 auf das Expositionsmuster am Arbeitsplatz siehe Nummer 4.2.
(3) Details zur Vorgehensweise bei diesem T25-Verfahren sind der zitierten Literatur (z. B. EC, 1999, oder auch REACH RIP 3.2-1B preliminary Technical Guidance Document) zu entnehmen. Die wichtigsten Punkte sind:
Als Ausgangspunkt wird die niedrigste Dosisgruppe gewählt, die eine signifikant erhöhte Tumorinzidenz aufweist.
Das Kriterium der Signifikanz ist entweder auf statistischer (Fischer Exact Test zum Vergleich der Dosis- mit der Kontrollgruppe) oder auf biologischer Basis festzulegen. Analog FDA (2001) wird die Verwendung eines Signifikanzniveaus von p < 0,05 für seltene Tumore bzw. Tumore mit einer Spontaninzidenz <= 10 % bzw. p < 0,01 für Tumore mit höherer Spontaninzidenz als 10 Prozent vorgesehen. Ggf. sind neben der experimentellen Kontrollgruppe auch die Daten der historischen Kontrolle vergleichend heranzuziehen (vgl. zu historischen Kontrollinzidenzen z. B. Derelanko und Hollinger, 2002).
Von der Tumorinzidenz in der behandelten Gruppe wird die Spontaninzidenz in der Kontrollgruppe abgezogen.
Eine Korrektur für aufgetretene Mortalität wird im Allgemeinen nicht vorgenommen, so dass bei hoher Mortalität in der betrachteten Dosisgruppe die damit verbundene erhöhte Unsicherheit des T25-Wertes zu diskutieren oder die nächst niedrigere Dosisgruppe zu wählen ist. Hohe Mortalität kann auch bedeuten, dass die Studie nicht mehr für eine Risikoquantifizierung herangezogen werden kann (siehe Nummer 7, Minimalkriterien).
T25-Werte werden in der Regel getrennt für Spezies, Geschlecht und Organ/Tumortyp berechnet (siehe Nummer 3.1 Abs. 6).
Eine Zusammenfassung von Tumortypen/Organen/Geschlechtern kann mit Begründung erfolgen (siehe Nummer 3.1 Abs. 6).
Eine gegenüber der Standard-Lebensspanne der Versuchsspezies verkürzte Expositionsdauer und verkürzte Expositionszeit wird korrigiert.
Die gegenüber der Standard-Lebensspanne (w in Wochen) der Versuchsspezies verkürzte Expositionsdauer (w1 in Wochen) und verkürzte Expositionszeit (w2 in Wochen) wird durch Multiplikation mit dem Faktor (w1/w) x (w2/w) korrigiert (siehe Nummer 4.4).
Gegenüber den gewählten Standardwerten abweichende Expositionsschemata werden berücksichtigt.
Dies erfolgt durch lineare Korrekturfaktoren etwa bei Dosierungen/Tag, Expositionstage/Woche, sowie Expositionsdauer/Tag bei Inhalation.
Für die Risikoquantifizierung wird der niedrigste, als humanrelevant (in Bezug auf Spezies/Organ/Tumortyp) erachtete T25-Wert verwendet (siehe auch Nummer 3.1).
Diese Ausführungen sind nicht in voller Übereinstimmung mit der üblichen Vorgehensweise nach EU: Der T25-Wert wurde ursprünglich als körpergewichtsbezogene Stoffdosis konzipiert und somit in mg/kg Körpergewicht/Tag angegeben. Bei Vorliegen mehrerer Studien, die nicht alle Schlundsondierung benutzten, sondern Tiere z. B. über Trinkwasser, Futter oder Atemluft exponierten, wird die Umrechnung der Exposition auf die körpergewichtsbezogene Dosis als gemeinsame Vergleichsbasis vorgeschlagen (EC, 1999). Im vorliegenden Fall ist jedoch die Ausweisung einer Konzentrationsangabe erforderlich (mg/m3).
Wenn keine Pfad-zu-Pfad-Übertragung zulässig ist (siehe Nummer 4.3), dann kann der entsprechende (orale oder dermale) Ausgangswert für einen inhalativen T25 nicht verwendet werden.
(4) Der T25 wird mit den Faktoren, wie in Nummer 4 spezifiziert, in ein Humanäquivalent umgerechnet (hT25).
3.7 Vorgehen im Falle vorliegender Humandaten
Die Einordnung der Rolle epidemiologischer Beobachtungsstudien im Vergleich zum Tierexperiment bei der Quantifizierung von Krebsrisiken am Arbeitsplatz erfolgte bereits in Nummer 1.1 und bei der Erläuterung der zu Grunde zu legenden Datenbasis (Nummer 1.5 Abs. 1). Zum hier verwendeten Risikobegriff wird auf Nummer 1.4 verwiesen (Risikozahl).
Die folgenden Hinweise zum Vorgehen setzen eine adäquate epidemiologische Datenlage voraus (für Mindestkriterien siehe Nummer 7.6 dieses Leitfadens).
(1) Bei der Auswahl epidemiologischer Studien ist wie folgt vorzugehen:
Die vorhandene Studienevidenz sollte mittels einer strukturierten, systematischen Literatursuche identifiziert und auf ihre Qualität und Eignung für die Risikobewertung geprüft werden. Prinzipien, die für die Auswahl von arbeitsepidemiologischen Studien zur Durchführung einer Meta-Analyse aufgestellt wurden, sollten hier berücksichtigt werden. Es ist im Einzelfall zu entscheiden, ob mehrere Studien für die Bewertung in einer Meta-Analyse zu einem gepoolten Schätzer zusammengefasst werden oder ob einzelne Studien separat bewertet werden, um eine Spannweite von möglichen Risikoszenarien angegeben zu können.
Literatur: Blair et al, 1995; Roller et al, 2006, Kap. 5.2
Generell sind analytische Studiendesigns mit individueller Expositionseinschätzung zur Risikobewertung auszuwählen. Sowohl Kohorten- als auch Fall-Kontroll-Studien können dabei zur Risikobewertung herangezogen werden.
Die in der Arbeitsepidemiologie verwendeten beobachtenden Studiendesigns lassen sich nach absteigendem Evidenzgrad wie folgt ordnen:
- 1.
Kohortenstudie;
- 2.
Fall-Kontroll-Studie (FKS);
- 3.
Querschnittstudie (QS);
- 4.
Ökologische oder Korrelationsstudie (siehe auch Glossar).
Quantitative Expositionsdaten stehen häufiger in Kohortenstudien zur Verfügung, während Fall-Kontroll-Studien in der Regel eine bessere Berücksichtigung von Störeinflüssen (Confounding) gewährleisten (weitere Details zu den besonderen Stärken und Schwächen der Studiendesigns siehe Ahrens et al, 2008). In begründeten Ausnahmefällen, z. B. im Falle einer in eine Kohorte eingebetteten Fall-Kontroll-Studie mit spezifischeren oder genaueren Informationen zu Exposition und/oder Endpunkt, kann eine FKS besser für eine Abschätzung von Arbeitsplatzgrenzwerten geeignet sein, als die zu Grunde liegende Kohortenstudie.
(2) Bei der Berücksichtigung von Zielparametern ist wie folgt vorzugehen:
Generell sind Maße mit Bezug zur Krebsinzidenz denen der Krebsmortalität vorzuziehen, es sei denn, Inzidenz und Mortalität können aufgrund einer hohen Letalität der Erkrankung (wie z. B. beim Lungenkarzinom) als identisch angesehen werden.
Je feiner die betrachteten Endpunkte aufgegliedert werden, umso geringer ist die zahlenmäßige Besetzung der Strata. Es ist also im Einzelfall abzuwägen, ob sich verschiedene Endpunkte sinnvoll kombinieren lassen, um die statistische Power zu erhöhen (d. h. Kombination verschiedener verwandter Tumorentitäten zu einer Gruppe), auch wenn sich kausale Faktoren im Einzelnen unterscheiden können, z. B. bei Leukämien und Lymphomen, Kopf-Hals-Tumoren usw.
Es ist im Einzelfall zu entscheiden, ob vorgezogene Endpunkte, wie z.B. biologische Marker, die als notwendige Vorstufe auf der Kausalkette zur untersuchten Zielerkrankung angesehen werden können, als Surrogatparameter in die Bewertung der Studienlage aufgenommen werden können. Ihre Einbeziehung ist sinnvoll, wenn die Demonstration eines frühen klinischen Effektes als Warnsignal anzusehen ist.
(Warnsignale können die Einführung von Schutzmaßnahmen rechtfertigen.)
(3) Für die Berechnung der Risikozahl kann folgendermaßen vorgegangen werden:
Ein Punktschätzer für jede Expositionskategorie (z.B. Median, geometrisches Mittel) ist die bevorzugte Angabe für die Risikoableitung.
Ist lediglich eine Expositionsrange berichtet worden (z.B. 1-9 ppm-Jahre), so kann für die Berechnung alternativ die Klassenmitte (hier 5 ppm-Jahre) zu Grunde gelegt werden. Konzentrationsangaben in mg/m sollten in stoffspezifische ppm umgerechnet werden. Für diese Berechnung zu Grunde gelegt werden dabei 240 Arbeitstage/Jahr und ein Atemvolumen von z. B. 10 m3 pro Arbeitstag, der mit acht Stunden veranschlagt wird (das Atemvolumen ist abhängig von der Arbeitsbelastung, 10 m3 betrifft z.B. eine leichte bis moderate Anstrengung)
(vgl. van Wijngaarden und Hertz-Picciotto (2004) bzw. Kap. 4.5 des Leitfadens).
Die kumulierten Konzentrationsangaben in ppm-Jahren sind danach auf den Langzeit-Mittelwert nach 40 Jahren umzurechnen.
Je nach Datenlage sind unmittelbar absolute Risikomaße (z.B. kumulatives Risiko) oder - wenn diese nicht berichtet wurden - Maße des Relativen Risikos zur Exposition in Beziehung zu setzen. In der Regel werden Maße wie SMR, SIR, RR oder OR vorliegen. Zur Berechnung des Lebenszeitrisikos der Exponierten können diese relativen Risikoerhöhungen mit einem Schätzwert für das Lebenszeitrisiko der Vergleichsgruppe, z. B. der Allgemeinbevölkerung, multipliziert werden, sofern nicht die ausführliche Sterbetafelmethode verwendet wird.
Das für die Expositionsspannweite berichtete Risikomaß (RR/SIR etc.) kann mit dem kumulierten Expositionswert in einer Regressionsanalyse korreliert werden, was eine Extrapolation in den Hoch- bzw. Niedrigrisikobereich und Aussagen zum Risiko pro Unit-Anstieg (1 ppm) der Exposition ermöglicht. Somit kann das Lebenszeitrisiko in Abhängigkeit von einer gegebenen Expositionshöhe bzw. einem angenommenen Arbeitsplatzgrenzwert geschätzt werden.
Nach Subtraktion des Risikos der Nicht-Exponierten (z.B. Allgemeinbevölkerung) wird ein Schätzwert des expositionsbezogenen Exzess-Risikos erhalten.
Einschränkungen der Aussagekraft der Ergebnisse sind zu diskutieren.
Es wird somit ein Vorgehen analog zu Roller et al. (2006) und Goldbohm et al. (2006) vorgeschlagen.
Einschränkungen der Aussagekraft der Ergebnisse sind z. B. Bias, mögliches residuelles Confounding, Misklassifikation usw. Die Verwendung von Risikoschätzern, die für Confoundereffekte adjustiert wurden, ist anzustreben. Wegen der Modellabhängigkeit der Adjustierung und zur Abschätzung der Stärke eines möglichen Confounding, sollten Berechnungen adjustierter vs. nicht adjustierter Risiken möglichst einander gegenübergestellt werden. Inkonsistente oder nicht vorhandene Dosis-Effektbeziehungen können in epidemiologischen Studien häufig beobachtet werden. Aber auch in den Fällen, in denen Studienergebnisse lediglich das Vorhandensein eines Ursache-Effektzusammenhangs andeuten, können die Daten berücksichtigt werden. Abweichungen von einer erwarteten Dosis-Effektbeziehung und ihre möglichen Ursachen und Konsequenzen für die Risikoextrapolation sind zu diskutieren.
Es ist zu bedenken, dass der vorgestellte Ansatz Variationen des Risikos zwischen Individuen aufgrund unterschiedlicher Suszeptibilität ignoriert. Die Übertragbarkeit der Ergebnisse auf andere Populationen muss in jedem Einzelfall evaluiert werden. Dabei sind auch mögliche Einschränkungen der Übertragbarkeit, z. B. bei Vorliegen eines Healthy Worker Effekts, zu berücksichtigen. Vor dem Hintergrund einer Bewertung des Risikos von Arbeitsstoffen und der Festlegung von Grenzwerten zur Verbesserung des Arbeitsschutzes sind diese Überlegungen jedoch von untergeordneter Natur. Bei semiquantitativen Expositionsangaben und sonst fehlenden epidemiologischen Daten kann versucht werden, ggf. durch Rückfrage bei den Autoren der Originalpublikationen Einstufungskriterien für Expositionsstufen zu ermitteln und dadurch eine quantitative Expositionsbewertung vornehmen zu können.
(4) Abweichungen vom Default sind in folgenden Fällen möglich:
Um die Konsistenz der Ergebnisse unter verschiedenen Voraussetzungen prüfen zu können, können von der kumulativen Exposition abweichende Expositionsmodelle (Intensität, Dauer, Expositionsspitzen, Wirkungsschwelle) je nach Wirkprinzip ebenfalls Berücksichtigung finden, falls entsprechende Schätzer in den bewerteten Artikeln dokumentiert wurden.
In Ermangelung einer adäquaten Studienlage kann im Ausnahmefall auf die Ergebnisse von Querschnitts- bzw. Korrelationsstudien zurückgegriffen werden. Die Aussagekraft derartiger Studienergebnisse ist mit großem Vorbehalt und unter ausführlicher Darstellung der Limitationen zu diskutieren. Querschnittstudien und ökologische Studien sollten in aller Regel bestenfalls als Ergänzung zu tierexperimentellen Daten herangezogen werden.
(5) Für die Extrapolation in den Niedrigrisikobereich wird auf das Vorgehen bei tierexperimentell-toxikologischen Daten verwiesen (siehe Nr. 5). Humandaten sollten nach Möglichkeit zur Überprüfung der Plausibilität der Extrapolationsfaktoren bei der Übertragung von Tierexperimenten auf den Menschen herangezogen werden.
4 Übertragung tierexperimenteller Daten auf den Menschen
4.1 Berücksichtigung von Speziesdifferenzen
(1) Für das Auftreten kanzerogener Wirkung wird bei der Ableitung von Risikozahlen in diesem Leitfaden in der Regel von gleicher Empfindlichkeit des Versuchstiers mit dem Menschen bei inhalativer Exposition ausgegangen. Diese Annahme ist nicht gut abgesichert; sie hat demnach bei nur beschränkter wissenschaftlicher Validierung Konventionscharakter.
Roller et al. (2006) zeigten für eine Reihe von Kanzerogenen bei Inhalationsstudien eine eher höhere Empfindlichkeit des Menschen im Vergleich zum Versuchstier. Die Autoren kamen damit zur Schlussfolgerung: "Die Ergebnisse deuten darauf hin, dass eine Speziesextrapolation anhand der äquivalenten Exposition ohne besondere Berücksichtigung toxikokinetischer und toxikodynamischer Speziesunterschiede in der Regel nicht zu einer Überschätzung des Risikos des Menschen führt." Mit diesem Befund lässt sich die Aussage in Nummer 4.1 Abs. 1 stützen. Roller et al. gehen jedoch weiter und schlagen auf Basis ihrer Befunde vor, auch dann gleiche Empfindlichkeit anzunehmen "wenn sich - zum Beispiel aufgrund mechanistischer Daten - eine geringere Empfindlichkeit des Menschen vermuten lässt."
(2) Stoffspezifische Angaben, die ein deutliches Abweichen vom Durchschnitt zeigen (z. B. aus pharmakokinetischen Modellen) können für die Begründung einer vom Default abweichenden Risikoquantifizierung herangezogen werden.
Dieses Vorgehen ermöglicht bei "deutlichem Abweichen vom Durchschnitt" ein Abweichen vom Default. Welches Gewicht an mechanistischen oder kinetischen Erkenntnissen eine geringere Empfindlichkeit des Menschen mit hinreichender Wahrscheinlichkeit vermuten lässt, ist eine Abwägung bzw. Einzelfallentscheidung ("expert judgement").
4.2 Vorgehen bei Vorliegen einer tierexperimentellen Inhalationsstudie
(1) Bei Substanzen mit einem Blut-Luft-Verteilungskoeffizienten > 10 und systemisch auftretenden Tumoren ist die im Tierexperiment eingesetzte Raumluftkonzentration (6h Exposition/Tag; Ruhebedingung) über einen Korrekturfaktor von 2 auf das Arbeitsplatzszenario (8h Exposition/Tag; leichte Aktivität) als humanäquivalente Expositionshöhe anzupassen.
Im Entwurf zum REACH-Implementation Projekt (REACH RIP 3.2 - 1B preliminary Technical Guidance Document) werden die Hintergrunddaten für diese Umrechnung erläutert:
| Ratte | Mensch | |
|---|---|---|
| Körpergewicht | 250 g | 70 kg |
| Atemvolumen (Standard; sRV) | 0,2 l/min/Ratte -> allometrisches Scaling* 0.8l/min/kg Körpergew. (KG) -> | 0,2 l/min/kg KG |
| | | | | | |
| v | v | v |
| Für verschiedene Expositionsdauer | ||
| 6 h Exposition | 0,29 m3/kg KG | 5 m3/Person |
| 8 h Exposition | 0,38 m3/kg bKG | 6,7 m3/Person |
| 24 h Exposition | 1,15 m3/kg KG | 20 m3/Person |
| Atemvolumen bei leichter beruflicher Aktivität (wRV) | ||
| 8 h Exposition | 10 m3/Person |
* Scalingfaktor 4 bei Ratte - Mensch
Danach entspricht bei systemischen Effekten z. B. ein T25 (Ratte) bei 6h Exposition/d von 10 mg/m3 einer hT25 (Mensch, 8h/Tag) von 5 mg/m3.
Da nicht für alle Stoffe der Blut-Luft-Verteilungskoeffizient bekannt ist, kann näherungsweise die Wasserlöslichkeit (> 1g/l, gut wasserlösliche Substanzen) herangezogen werden.
(2) Im Falle von Speziesunterschieden in der Resorption sind diese bei der Interspeziesextrapolation zu berücksichtigen.
4.3 Vorgehen bei Vorliegen einer tierexperimentellen Studie mit oraler Applikation
(1) Wenn keine studienspezifischen Informationen zur körpergewichtsbezogenen Dosis vorliegen, sondern nur Konzentrationen im Futter oder Wasser berichtet sind, können folgende Standardwerte zur Umrechnung verwendet werden (nach REACH RIP 3.2 - 1B preliminary Technical Guidance Document).
| Default values for body weights, food and-water intake for the calculation of doses in lifetime studies | ||||||
|---|---|---|---|---|---|---|
| Experimental animal | Sex | Body weight (kg) | Food consumption per day** (g) | Water consumption per day** (ml) | ||
| Mouse | Male | 0.03 | 3.6 | (120) | 5 | (167) |
| Female | 0.025 | 3.25 | (130) | 5 | (200) | |
| Rat | Male | 0.5 | 20 | (40) | 25 | (50) |
| Female | 0.35 | 17.5 | (50) | 20 | (57) | |
| Hamster | Male | 0.125 | 11.5 | (92) | 15 | (120) |
| Female | 0.110 | 11.5 | (105) | 15 | (136) | |
** In brackets the daily food or water consumption is given in g or ml per kg body weight per day, as appropriate.
(2) Eine im Tierexperiment applizierte Dosis (Einheit: mg/ kg Körpergewicht x Tag) wird in eine humanäquivalente Dosis durch Berücksichtigung eines allometrischen Scalingfaktors transformiert. Im Default wird hierzu die Umrechnung über das allometrische Scaling nach Grundumsatz vorgenommen ((KörpergewichtMensch / KörpergewichtTier)0,25). Es ergeben sich gerundet Faktoren von
Hund, Affe: 2
Ratte: 4
Maus: 7
Die Berücksichtigung eines Scalingfaktors bei zu Grunde liegender Oralstudie ist kein konservativer Extrapolationsschritt, sondern stellt im Standardfall eine biologisch begründete Datenanpassung dar (vgl TGD, Abschnitt 4.14.2.4, auch Tabelle 11 (Scalingfaktoren bei Standardgewichten); EPA, 2005; Kalberlah und Schneider, 1998).
(3) Die humanäquivalente Dosis ist im Folgeschritt in eine Luftkonzentration umzuwandeln, sofern nicht bestimmte Gründe gegen eine "Pfad-zu-Pfad-Extrapolation" sprechen: Gegen eine solche Umwandlung sprechen insbesondere:
ausgeprägter First Pass-Effekt,
lokale Tumoren im Respirationstrakt zu erwarten (besonders bei lokal wirksamen aber auch persistenten Stoffen sowie Metallverbindungen relevant),
lokale Tumoren bei oraler Applikation spielen eine bewertungsrelevante Rolle (z. B. Vormagentumoren im Nager),
deutlich abweichende Organkonzentration im kritischen Zielorgan sind bei Inhalation zu erwarten und bewertungsrelevant (z. B. bei Studien mit Schlundsondenapplikation oft maßgeblich).
Differierende Pfad-spezifische Resorptionsquoten sind bei einer Pfad-zu-Pfad-Extrapolation zu korrigieren.
Die Grenzen der Pfad-zu-Pfad-Extrapolation wurden z. B. bei der Erarbeitung des ARW-Konzepts im Ausschuss für Gefahrstoffe ausgewiesen. Vgl. AKW-Konzept (o.V, 1998)
(4) Ist bei einer Studie mit oraler Applikation keine Pfad-zu-Pfad-Extrapolation möglich und liegen keine Inhalationsstudien oder Erkenntnisse mit inhalativer Aufnahme des Kanzerogens beim Menschen vor, ist in der Regel keine Risikoquantifizierung möglich (siehe Nummer 7).
4.4 Vorgehen bei Studien mit verkürzter Expositions- und/oder Beobachtungsdauer
(1) Wurde die Exposition vor Ende des Experiments gestoppt (längere Nachbeobachtungszeit), so ist eine Korrekturrechnung vorzunehmen. Bei einer angenommenen experimentellen Spanne von 100 Wochen bedeutet das zum Beispiel:
tatsächliche Exposition: 70 Wochen lang 50 ppm im Futter, 30 Wochen Nachbeobachtung:
kalkulierte Exposition: 50 ppm x 70/(30 + 70) = 35 ppm für die gesamte experimentelle Spanne.
Wenn alle Tiere einer Dosisgruppe vorzeitig sterben, wird die Expositions- und Lebenszeit des langlebigsten Tiers für die Umrechnung zu Grunde gelegt.
(Quelle: Swirsky Gold et al.,http://potency.berkeley.edu)
Wenn eine Expositionsdauer von ca. 100 Wochen im Tierversuch in ein Humanäquivalent umgerechnet wird, übersteigt dieses Äquivalent die anteilige Lebensarbeitszeit von ca. 40 Jahren. Auch wenn in weiteren Schritten von Lebenszeitexposition auf Exposition über Lebensarbeitsdauer rückgerechnet wird, stellt es also einen konservativen Ansatz dar, die Beobachtungen nach dieser längeren Expositionsspanne für die Quantifizierungen zu Grunde zu legen.
(2) Ist die Experimentspanne kleiner als die Lebensspanne, erfolgt in der Regel eine weitere Korrektur von Experimentalspanne auf Lebensspanne mit Korrekturfaktor f2, mit f = Experimentalspanne/Standard-Lebensspanne (Bsp. Experiment nach 100 w beendet, Standard-Lebensspanne 104 w: Korrekturfaktor = (100/104)2 = 0,92). Als Standard-Lebensspannen werden angenommen: Maus, Ratte, Hamster: 2 Jahre, Hund: 11 Jahre, Affe (Macaca): 20 Jahre.
Auch Dybing et al. (1997) wählen bei ihrem T25-Konzept einen entsprechenden Ansatz (siehe auch Nummer 3.6 Abs. 3 dieses Leitfadens):
Verkürzte Exposition (w1) gegenüber der Gesamtversuchsdauer (w2 Wochen):
Korrekturfaktor f = w1/w2
Verkürztes Experiment (w1) gegenüber Gesamtlebensspanne (w2 Wochen):
Korrekturfaktor f = (w1/w2)2
Bei dieser "Standard-Lebensspanne" handelt es sich um eine wenig konservative Konvention. Abweichend von diesem Standard kann es insbesondere bei Lungentumoren (Ratten) erforderlich sein, eine verlängerte Lebensdauer anzunehmen. Bei der Ratte treten expositionsbedingte Lungentumoren vor allem im Alter von mehr als zwei fahren auf. Die Spontanrate für Lungentumoren ist bei Ratten niedrig, nach 2,5 Jahren um ein bis zwei Prozent, beim einen Stamm etwas höher, beim anderen Stamm sogar noch niedriger. Die Beobachtungszeit sollte für die quantitative Risikoabschätzung dort unbedingt mehr als zwei Jahre betragen. Es heißt z. B. bei McConnell und Swenherg (1994): "Following the 24-mo exposure period, the animals were held for lifetime Observation (until ~20 Prozent survived)". Dies impliziert, dass 24 Monate keine Lebenszeit-Beobachtung sind, dass aber aus pragmatischen Gründen ein bestimmtes Kriterium (hier 20 Prozent Überlebensquote) zur Definition der "Lebenszeit" (größer als 24 Monate) verwendet werden kann.
(3) Im Falle einer Absenkung der Expositionskonzentration während des Versuchs wird in der Regel das zeitgewichtete Mittel für die Expositionshöhe herangezogen.
Der einfache Ansatz eines kumulativen Dosismaßes über die gesamte Lebenszeitspanne (nach Druckrey, siehe unten) berücksichtigt nicht, dass ein krebserzeugender Stoff spezifisch eine oder mehrere Stufen der Kanzerogenese auszulösen vermag. Wenn ein frühes Stadium der Kanzerogenese betroffen ist, sind Expositionen am Anfang des Lebens besonders kritisch. Persistierende Substanzen können auch nach einem frühen Abbruch der Behandlung eine anhaltende innere Belastung aufrechterhalten.
Die "Guidelines for Carcinogen Risk Assessment" (2005) der U.S. EPA geben zu bedenken (http://www.epa.gov/IRIS/cancer032505.pdf): "For chronic exposure studies, the cumulative exposure or dose administered often is expressed as an average over the duration of the study, as one consistent dose metric. This approach implies that a higher dose administered over a short duration is equivalent to a commensurately lower dose administered over a longer duration. Uncertainty usually increases as the duration becomes shorter relative to the averaging duration or the intermittent doses become more interne than the averaged dose. Moreover, doses during any specific susceptible or refractory period would not be equivalent to doses at other times. For these reasons, cumulative exposure or potential dose may be replaced by more appropriate dose metric when indicated by the data."
Zum Multistage- und Moolgavkar-Modell gibt es beispielsweise mathematische Anpassungsvorschläge für intermittierende und Kurzzeit-Expositionen in beliebigen Lebensabschnitten (Crump und Howe, 1984; Chen et al, 1988; Yamasaki, 1988). Diese erscheinen aber zu komplex für die routinemäßige Anwendung.
Nach der Druckrey'schen Regel ist die Tumorgenität einer der im Laufe des Lebens einwirkenden Gesamtdosis (d x t = const.). Diese Beschreibung hat für viele gentoxische Stoffe Gültigkeit. Sie berücksichtigt allerdings keine Depoteffekte, d. h. konstante Einwirkungen schwerlöslicher oder anderweitig biopersistenter Stoffe nach Inhalation oder Injektion (wie z. B. Metallverbindungen, Asbest, Holzstaub). Die Druckrey'sche Regel kann auch die Spätfolgen kurzfristig einwirkender hoher, gewebsschädigender Dosen unterschätzen, etwa weil gesteigerte Zellproliferationsraten die Empfindlichkeit der Zielgewebe steigern, gentoxische Läsionen fixieren und Einwanderung von Stammzellen in Zielgewebe begünstigen. Die Druckrey'sche Regel ist jedoch die primäre Grundlage der linearen Dosisextrapolation und auch der üblichen Zeitextrapolation.
Literatur: Chen et al. (1988); Yamasaki (1988); Crump und Howe (1984); Dybing et al. (1997)
(4) Experimente, bei denen die Expositionszeit weniger als die Hälfte der Standard-Lebensspanne dauern, sind für eine Risikoquantifizierung nicht geeignet. Die Beobachtungsdauer in einem Versuch mit Mäusen sollte in der Regel nicht unter 18 Monaten liegen, in einem Versuch mit Ratten nicht unter zwei Jahren.
In grober Annäherung entspricht die Hälfte der Standard-Lebensspanne etwa dem Verhältnis zwischen Lebensdauer und Lebensarbeitszeit beim Menschen. Damit ist z. B. eine Expositionsdauer von einem Jahr (Ratte) in der Regel ausreichend, um die entsprechenden Tumorbefunde quantitativ verwerten zu können. Ist jedoch die Nachbeobachtungszeit kurz, sind relevante Risikounterschätzungen zu befürchten.
4.5 Normierung der täglichen Expositionsdauer
(1) Für die Exposition am Arbeitsplatz gelten folgende Standardannahmen: Expositionsdauer während des Arbeitslebens 40 Jahre, Dauer des Arbeitstags: 8 Stunden, Wochenarbeitszeit: 5d/Woche, Arbeitswochen/Jahr: 48 Wochen; Körpergewicht: 70 kg, Atemvolumen: 10 m3 /Arbeitstag (8 h). Eine Umrechnung vorliegender abweichender Expositionsmuster auf die hier referierten Standardannahmen erfolgt in der Regel linear. Liegen Erkenntnisse aus der Allgemeinbevölkerung vor, so werden bei dieser (wenn nicht anders angegeben) folgende Expositionsparameter unterstellt: Expositionsdauer 75 Jahre, Körpergewicht: 70 kg, Nahrungsaufnahme/Tag 1,4 kg, Wasseraufnahme: 2 Liter/Tag, Atemvolumen: 20 m3 /Tag (24 h).
Zur Umrechnung auf Basis des Tierexperiments ist darauf zu achten, dass keine Doppelberechnung erfolgt: Nach Nummer 4.2 Abs. 1 erfolgt bei wasserlöslichen Substanzen bereits eine Umrechnung von 6h/d (Ruhebedingungen, Tierexperiment) auf 8h/d (leichte Aktivität; Arbeitsplatz) über einen Faktor 2.
(2) Bei Extrapolation vom Tierexperiment auf den Menschen ist in der Regel die experimentelle Expositionsdauer (pro Tag/pro Woche) angegeben und wird linear auf die oben genannte Zeitdauer (berufliche Exposition) umgerechnet.
Dieser Ansatz geht von der biologischen Modellannahme aus, dass die kumulative Dosis (c x t) einer Einwirkung das Risiko bestimmende Dosismaß darstellt. Dieses Vorgehen wird (für den Standardfall) gewählt, obwohl bekannt ist, dass es sich hier meist um einen konservativen Vereinfachungsschritt handelt. Die Parameter wurden in ihrer Höhe vom Technical Guidance Document der EU übernommen (vgl. dort Abschnitt 4.14.2.5 und Tabelle 12).
5 Extrapolation auf niedrigere Risikohöhen
5.1 Festlegung des Vorgehens nach dem Wirkprinzip
(1) Wurde nach den Erkenntnissen in Nummer 2 ein im Wesentlichen durch die direkte Gentoxizität determiniertes Wirkprinzip für die Kanzerogenese festgestellt, so erfolgt im Standardfall eine lineare Extrapolation.
(2) Wurde nach den Erkenntnissen in Nummer 2 festgestellt, dass das Wirkprinzip alleine durch nichtgentoxische Ereignisse geprägt ist und kann für den oder die bestimmenden Parameter eine Dosis-Wirkungsbeziehung mit einer Wirkungsschwelle benannt werden, so ist diese zu berechnen.
(3) Ist kein Wirkprinzip bekannt bzw. ausreichend gesichert, erfolgt im Standardfall ebenfalls eine lineare Extrapolation.
(4) In Fällen, wo das Wirkprinzip zwar im Wesentlichen bekannt ist, jedoch
- 1.
eine direkte Gentoxizität keine dominierende Bedeutung besitzt,
- 2.
keine eindeutige Wirkungsschwelle für die Kanzerogenität vorliegt oder
- 3.
eine Schwelle aufgrund der Datenlage nicht quantifiziert werden kann,
wird im Regelfall ein sublinearer Dosis-Wirkungsverlauf in den Niedrigrisikobereich unterstellt.
Zum Begriff der "Wirkungsschwelle" sind die Ausführungen in Nummer 2.5 zu beachten. Grundsätzlich ist ein NOAEL für kanzerogene Effekte (keine beobachtete signifikant erhöhte Inzidenz gegenüber Hintergrund) quantitativ nicht mit einer Schwelle gleichzusetzen.
(5) In Zweifelsfällen über die Zuordnung zu Absatz 1 -4 ist über parallele Risikoquantifizierungen nach verschiedenen Methoden (siehe Nummer 5.2) zu prüfen, ob sich Unterschiede ergeben und wie relevant die Festlegung auf ein Wirkprinzip ist. Ggf. ist bei nahe zusammenliegenden Dosis-Risikoverläufen auf eine Entscheidung zum vorherrschenden Wirkprinzip nicht notwendig, um ohne relevanten Fehler eine Risikoquantifizierung vorzunehmen. Die Unsicherheit in der Risikoquantifizierung ist zu dokumentieren. Für den Fall, dass die parallelen Risikoquantifizierungen zwar für Expositionen mit erhöhtem Risiko noch zu vergleichbaren Risikozahlen führen (z.B. bei zusätzlichen Lebenszeitrisiken bis in den Promillebereich), jedoch bei niedrigeren Risiken gravierende Abweichungen auftreten, ist der Gültigkeitsbereich entsprechender Dosis-Risiko-Verläufe abzugrenzen.
(6) Eine Risikoextrapolation in den Niedrigrisikobereich unter Verwendung derjenigen Modellfunktion, die für den experimentellen Bereich die beste Anpassung an die Daten gezeigt hat, ist in der Regel kein geeignetes Vorgehen. Es ist z. B. möglich, dass im experimentellen Bereich Supralinearität vorliegt, jedoch Sublinearität im Niedrigrisikobereich.
Die Konvention, die Benchmark-Methode bei belegter Sublinearität als Ersatz für das linearisierte Multistage-Modell als mechanistisch begründet für den experimentellen Bereich und den Niedrigrisikobereich zu verwenden (siehe Nummer 3.2 Abs. 3 und Nummer 5.2 Abs. 3), widerspricht dieser Aussage Absatz 6. Diese Modellierung wird jedoch deshalb als für die Extrapolation herangezogen, weil sie eine einfache Konvention zur Beschreibung einer Sublinearität bietet. Es ist jedoch nicht daraus zu schlussfolgern, dass mit diesem Modell die "richtige" Steigung im Niedrigrisikobereich gefunden wurde.
5.2 Extrapolation auf niedrigere Risikohöhen bei nichtlinearem Verlauf
(1) Es wird eine Informationslage entsprechend Fall Absatz 4 in Nummer 5.1 unterstellt, so dass mit hinreichender Wahrscheinlichkeit von einem nichtlinearen Dosis-Wirkungsverlauf auszugehen ist. In diesem Fall wird eine plausible Festsetzung für diese nichtlineare Funktion vorgenommen.
(2) Ist die Datenlage hinreichend qualifiziert, dass das Benchmark-Verfahren eingesetzt werden kann, dann wird unterstellt, dass mit der Benchmark-Modellierung auch die Nichtlinearität in einem Risikobereich >= 1:1.000 abgebildet werden kann, auch wenn der experimentelle Bereich nur Risiken ab z. B. 1 Prozent oder ab 5 Prozent abdeckt. Zwischen BMD0,1 (1:1.000) und Ursprung bzw. Hintergrund wird linear extrapoliert.
Der Bezug zur BMD statt zur BMDL ist deshalb gerechtfertigt,
- 1.
weil es sich bei der Orientierung an der BMD um die Schätzung mit der höchsten 'Wahrscheinlichkeit handelt ("maximum likelihood"),
- 2.
weil nach Absatz 4 in Nummer 5.1 zusätzliche inhaltliche Gründe vorliegen müssen, die einen nichtlinearen Verlauf stützen, so dass Modellierungen, die über die BMDL mathematisch als möglich angesehen werden müssten, aus diesen inhaltlichen (z. B. mechanistischen) Gründen als unwahrscheinlich angesehen werden,
- 3.
weil aufgrund der Qualitätskriterien Benchmark-Modellierungen nur dann als adäquat betrachtet werden, wenn die Unterschiede zwischen BMD und BMDL gering sind, so dass nicht mit einer relevanten Risikounterschätzung bei Bezug auf die BMD zu rechnen ist (selbst wenn "in Wirklichkeit'" die BMDL das Risiko korrekter widerspiegelt sollte). Ferner ergibt sich das Vorgehen aus der methodische Kontinuität zum T25, der ebenfalls keinen Vertrauensbereich enthält.
Die folgenden Beispiele (Fall A, B) zeigen eine Abgrenzung zwischen einem Fall mit Nichtlinearität (Fall A) und Linearität (Fall B). In Fall A wären zusätzliche mechanistische Hinweise erforderlich, die die Nichtlinearität stützen. Können diese nicht gegeben werden, stellt die BMD10 den POD dar, unterhalb dessen eine lineare Extrapolation erfolgen würde.
(3) Wurde der T25 als POD für das Krebsgeschehen herangezogen, dann wird für den Fall begründeter Nichtlinearität angenommen, dass eine nichtkanzerogene Wirkung als Verstärkungsmechanismus (z. B. Reizung im Respirationstrakt, Zytotoxizität in der Niere), die zum Krebsgeschehen in höherer Dosierung maßgeblich beiträgt, quantitativ beschrieben werden kann. Die Ermittlung des anzunehmenden Expositions-Risikoverlaufs erfolgt dann in vier Schritten.
| Schritt 1: | Für diese (für sich nichtkanzerogene) verstärkende Wirkung wird eine humanäquivalente Wirkungsschwelle (TC*; als Konzentration in der Luft) ermittelt, indem übliche Extrapolationsfaktoren berücksichtigt werden. |
|---|---|
| Extrapolationsverfahren für nichtkanzerogene Wirkungen werden nach dem Ansatz der EU (DNEL; RIP 3.2.2) durchgeführt. | |
| Schritt 2: | Es wird - ausgehend vom normalisierten und als Humanäquivalent umgerechneten T25 (hT25) - als Zwischenrechnung das Krebsrisiko (10-p) bei linearer Extrapolation zwischen T25 und Ursprung bzw. Hintergrund am Punkt TC* berechnet. |
| Schritt 3: | Dem Punkt TC* wird dann pragmatisch ein zehnfach geringeres Krebsrisiko (1 Größenordnung: 10-(p-1)) als bei linearer Extrapolation zugewiesen. |
| Schritt 4: | Schließlich wird vom Punkt TC* linear zum T25 und zum Ursprung (bzw. zum Hintergrund) linear extrapoliert. Das nominelle Risiko kann somit für jeden Punkt zwischen Nullpunkt und T25 genannt werden mit einer Knickstelle der Funktion bei der extrapolierten Wirkungsschwelle (TC*) für den Verstärkungsmechanismus. |
| Die Vorgehensweise bei diesem "Hockey stick"-Ansatz berücksichtigt, dass in der Regel zwar bekannt ist, wenn ein nichtlinearer Verlauf für die Konzentrations-Risiko-Beziehung zu unterstellen ist, dass jedoch weitere Parameter, die die Nichtlinearität des Krebsgeschehens quantitativ beschreiben, nicht bekannt sind. Der unbekannte Grad des "Durchhängens" der sublinearen Funktion wird durch einen Abschlagsfaktor an der extrapolierten Wirkungsschwelle ersetzt. | |
| Die folgende Abbildung zeigt prinzipiell die oben genannten Schritte in dem Fall, dass für das Krebsgeschehen ein T23 vorliegt und zusätzlich genügende Daten vorliegen, um für einen Verstärkermechanismus eine Wirkungsschwelle (TC*) zu ermitteln (Erläuterung siehe Text): |
Sublinearität bei Gentoxizität plus Verstärkermechanismus
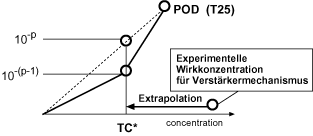
Bei der Berechnung des T25 sind zuvor die in den Nummern 3.6, 4.2, 4.4 erforderlichen Normierungen vorzunehmen.
FALL A: Gute Datenlage verweist auf nichtlineare Verhältnisse
| Konzentration (mg/m3) | Anzahl | Anzahl | Kommentar: |
|---|---|---|---|
| Tiere | Tumoren | ||
| 0 | 50 | 0 | Verlauf spricht für eine |
| 10 | 50 | 1 | deutliche Nichtlinearität; |
| 50 | 50 | 0 | gute Datenlage; z. B. |
| 200 | 50 | 10 | mechanistische Hinweise auf |
| 1.000 | 50 | 45 | Nichtlinearität |
Ergebnis, graphisch:
Ergebnis in Zahlen, Erläuterung:
| Modell | BMD10 | BMDL10 | BMDL0,1 = 1 Promille | BMD0,1 = 1 Promille | T25 | T25/250 = 1 Promille |
|---|---|---|---|---|---|---|
| 150 | 110 | 22 | 40 | 235 | 0,94 | |
| Kommentar 1: | Unterschied 40/0,94 zeigt, dass BMD (1 Promille) deutlich geringeres Risiko ausweist als der bei dieser Datenlage nicht geeignete T25-Ansatz | 40 | <- -> | 0,94 | ||
| Kommentar 2: | Der geringer Unterschied zwischen 150 und 110 (bzw. 40 und 22) zeigt, dass kein relevanter Unterschied zwischen BMD und BMDL bei guter Datenlage besteht | |||||
| Kommentar 3: | Es wurde mit Logprobit modelliert. Wegen AIC=100,87 und p-Wert:0,34, chi-square=2,15 ist dies gegenüber Multistage gerechtfertigt. Dort: AIC: 103, p-Wert:0,19; Chi-Square: 3,3 und somit schlechtere Anpassung -> Multistage würde Nichtlinearität kaum zeigen | |||||
FALL B: Mittlere Datenlage lässt nichtlineare oder lineare Verhältnisse zu
| Konzentration (mg/m3) | Anzahl | Anzahl | Kommentar: |
|---|---|---|---|
| Tiere | Tumoren | ||
| 0 | 50 | 0 | Verlauf schließt Nichtlinearität nicht |
| 10 | 50 | 1 | aus, doch auch Linearität möglich; mittlere Datenlage (Kriterien nach |
| 200 | 50 | 10 | Leitfaden 3.1 erfüllt) |
| 1.000 | 50 | 45 |
Ergebnis, graphisch:
Ergebnis in Zahlen, Erläuterung:
| Modell | BMD10 | BMDL10 | BMDL0,1 = 1 Promille | BMDL0,1 = 1 Promille | T25 | Linear: T25/250 = 1 Promille |
|---|---|---|---|---|---|---|
| 99 | 58 | 0,56 | 1,1 | 231 | 0,92 | |
| Kommentar 1: | Unterschied 0,92/1,1 zeigt, dass BMDL (1 Promille) fast identisches Risiko ausweist wie T25-Ansatz, da Linearität möglich (siehe Graphik) | 1,1 | <- -> | 0,92 | ||
| Kommentar 2: | Der Unterschied zwischen BMD und BMDL ist nicht erheblich | |||||
| Kommentar 3: | Es wurde mit Multistage (2 Freiheitsgrade) modelliert. Wegen AIC=98,86 und p-Wert:0,43, chi-square=0,63 ist dies gegenüber Logprobit gerechtfertigt. Dort: AIC: 99,74 p-Wert:0,31; Chi-Square: 1,01 und somit schlechtere Anpassung -> ähnliche Extrapolation linear/ Benchmark-Verfahren | |||||
Ein Beispiel zur Berechnung ist im Anhang (Nummer 10.2) aufgeführt.
5.3 Extrapolation bei angenommenem Schwellenphänomen
(1) Wird eine Mindestdosis bzw. Wirkungsschwelle für die Kanzerogenese angenommen (Absatz 2 in Nummer 5.1), so ist die Schwellendosis auf Basis vorliegender experimenteller Daten unter Einschluss bestimmter Extrapolationsfaktoren zu quantifizieren. Es wird vorausgesetzt, dass in diesem Falle weder direkte Gentoxizität und andere Wirkprinzipien ohne Schwelle eine Rolle spielen.
(2) Für die Festlegung der Schwellendosis sind besonders sorgfältig gerade auch Frühzeichen der entsprechend relevanten kritischen Veränderung zu erfassen, z. B. wären bei krebsrelevanter Nephrotoxizität auch erste Frühschäden in der Niere, die sich z.B. durch entsprechende Eiweißausscheidungen manifestieren, einzubeziehen. Dosis-Wirkungsbeziehung, LOAEL und NOAEL für diese (selbst nicht krebserzeugende, jedoch) für die Krebsentstehung als maßgeblich angesehene Wirkung sind zu ermitteln.
(3) Liegen keine entsprechend differenzierten Studienbefunde zu frühen Schädigungen vor, die als maßgeblich für die krebserzeugende Wirkung angesehen werden, soll dies über konservative Extrapolationsfaktoren ausgeglichen werden. In diesem Sinne erfordert die Festlegung z. B. einer Reizschwelle für einen nicht krebserzeugenden Stoff niedrigere Extrapolationsfaktoren als die Festlegung einer Reizschwelle bei einem Stoff, bei dem Reizung ein wichtiger Parameter für das Wirkprinzip bei Krebs darstellt.
(4) Aus diesem Grunde wird eine Erweiterung der üblichen Extrapolationsfaktoren um den Faktor 10 vorgenommen, so dass auf dem Hintergrund des möglichen Folgeeffekts Krebs die (zu unterschreitende) Wirkungsschwelle besonders sicher abgeschätzt wird. Nach der Terminologie in Nummer 5.2 liegt damit diese konservative Wirkungsschwelle bei TC*/10, wobei TC* sich dann nicht auf krebsverstärkende, sondern auf krebsauslösende Wirkungen bezieht.
Extrapolationen zur Berechnung von TC* verlaufen entsprechend der DNEL-Kalkulation (RIP 3.2.2).
Versteht man den "üblichen" NOAEL als einen Wert, der durchaus noch mit einem Effektniveau von fünf Prozent verbunden sein kann (auch wenn im experimentellen System keine Wirkung mehr beobachtet wird), so wird über diesen Faktor 10 ein deutlich kleineres Effektniveau mit dem resultierenden NAEL zu verbinden sein (z. B. Effektniveau 0,5 Prozent).
Das Vorgehen deckt sich mit dem Verständnis der einzelnen Extrapolationsfaktoren als bestimmtes Perzentil einer Verteilung (z. B. 90-Perzentil beim Intraspeziesfaktor): die Wahl eines zusätzlichen Extrapolationsfaktors ist gleichbedeutend mit der Erhöhung z. B. des Intraspeziesfaktors zum Einschluss eines höheren Perzentils (z. B. 95-Perzentil) verschiedener Empfindlichkeiten, wird aber pauschal einbezogen (nicht auf einen Einzelfaktor wie Intraspeziesfaktor oder Interspeziesvariabilitätsfaktor oder Zeitfaktor bezogen, sondern auf Gesamtverteilung, d. h. multiplizierte Einzelfaktoren).
(5) In Verbindung mit dem Benchmark-Verfahren für Krebsrisiken wird der Risikoverlauf entlang der modellierten Funktion (als BMD) bis zum Risiko bei einem Prozent angenommen. Damit wird vorausgesetzt, dass die Qualitätsmaßstäbe zur Anwendung des Benchmark-Verfahrens eingehalten sind (siehe Nummer 3.3). Mechanistische Erkenntnisse dürfen dem modellierten Verlauf der Expositions-Risiko-Beziehung nicht widersprechen. Ein Risiko "Null" wird dann pragmatisch bei einer BMD01/10 angenommen.
Für die Quantifizierung der Expositions-Risiko-Beziehung im Bereich oberhalb der angenommenen Wirkungsschwelle erfolgt demnach im vorliegenden Leitfaden nur dann eine Vorgabe, wenn eine Benchmark-Modellierung erfolgte. Liegt keine Benchmark-Modellierung vor, wird die Wirkungsschwelle nach Nummer 5.3 Abs. 4 berechnet, jedoch keine allgemeine Aussage zum Verlauf der Expositions-Risiko-Beziehung oberhalb dieser Wirkschwelle gemacht (ggf. ist eine Einzelfallbetrachtung erforderlich).
Für den Fall, dass das Krebsgeschehen qualifiziert in einer Benchmark-Modellierung abgebildet werden kann, ergibt sich folgende Darstellung für das Extrapolationsverfahren. Die errechnete Wirkungsschwelle (BMD01/10) ist vor ihrer regulatorischen Anwendung noch auf ein Humanäquivalent (Arbeitsplatzszenario) umzurechnen.
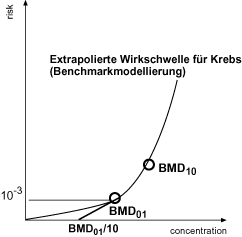
6 Intraspeziesextrapolation
6.1 Verzicht auf Intraspeziesextrapolation
(1) Es wird keine Intraspeziesextrapolation durchgeführt. Der Blickwinkel liegt demnach auf dem durchschnittlichen individuellen Risiko als zusätzliches Lebens(arbeits)zeitrisiko. Dennoch wird auch der Schutz der empfindlichen Personengruppen indirekt berücksichtigt, indem ein niedrigeres durchschnittliches Risiko beim Risikomanagement herangezogen werden soll (so dass auch das Risiko für empfindliche Personengruppen niedriger wird). Während bei Nichtkanzerogenen der (weitgehende) Schutz der Empfindlichen vor Gesundheitseffekten durch einen Intraspeziesfaktor explizit (als Default-Faktor für Variabilitäten) erfolgt, wird im Rahmen dieses Leitfadens also dieser Schutz bei Kanzerogenen durch die Auswahl eines entsprechend niedrigeren (als akzeptabel oder tolerabel erachteten) durchschnittlichen individuellen Risikos vorgeschlagen. Könnte man einen geeigneten Intraspeziesfaktor für krebserzeugende Wirkung beziffern, wäre eine direkte Umrechnung (auf das Risiko für empfindliche Personengruppen) möglich.
Die Nichtberücksichtigung des Intraspeziesfaktors bei Kanzerogenen entspricht einer häufiger angewandten Konvention. Es liegen nur unzureichende Daten vor, die bei diesem multifaktoriellen Geschehen die Spannbreite der Empfindlichkeiten hinreichend abbilden würden.
Es ist auf absehbare Zeit nicht erkennbar, dass für die Ausweisung eines wissenschaftlich gestützten Default-Werts für die Intraspeziesvariabilität hinreichende Daten aus dem Bereich krebserzeugender Wirkung vorliegen werden. Die Höhe eines entsprechenden Faktors wäre also äußerst unsicher. Eine vorläufige Auswertung tierexperimenteller Daten zeigte keine deutlich größere Variabilität von Auszuchtstämmen im Vergleich zu Inzuchtstämmen in Bezug auf das Krebsgeschehen. Eine einfache Verknüpfung von Enzymaktivitäten und deren Variabilität mit der Variabilität im Krebsgeschehen ist nicht möglich.
Einzelne Ansätze, vgl. z. B. EFSA (siehe auch Nummer 1.4 Abs. 3), weisen jedoch einen Intraspeziesfaktor von 10 auch für Kanzerogene aus, ohne dass sich dies bei diesem Vorschlag auf das Schutzniveau - Höhe des vorgesehenen Grenzwerts - auswirken würde. Bei EFSA wird unterstellt, dass die Intraspeziesvariabilität für krebserzeugende Effekte identisch mit derjenigen für andere Effekte wäre.
Auch von der U.S. EPA wird ein Intraspeziesfaktor für Krebs berücksichtigt, jedoch ausdrücklich nur für Kleinkinder, bei denen eine besondere Empfindlichkeit gezeigt ist, die in den tierexperimentellen Kanzerogenitätsstudien in der Regel nicht abgebildet wird. Das entsprechende Schutzgut kindliche Gesundheit ist in der vorliegenden Betrachtung nicht maßgeblich.
Für die Quantifizierung von nichtkanzerogenen Effekten, die als Auslöser oder Verstärker für Kanzerogenität berücksichtigt werden, werden jedoch empfindliche Personengruppen explizit berücksichtigt (siehe Nummer 5.2 Abs. 3 und Nummer 5.3 Abs. 4 dieses Leitfadens).
7 Minimalkriterien für eine Risikoquantifizierung
7.1 Einstufung der zu bewertenden Substanz
(1) Für Kanzerogene, die in die Kategorien 1 oder 2 (EU) für Kanzerogenität eingestuft sind, sollten in der Regel quantitative Abschätzungen der Expositions-Risiko-Beziehung vorgenommen werden.
(2) Zusätzlich sind in Einzelfallabwägung auch Substanzen mit Einstufung in Kanzerogenitätskategorie 3 bewertbar, insbesondere wenn diese Einstufung nicht durch die Qualität der Studie oder der Berichterstattung begründet ist und nicht durch die fragliche Humanrelevanz, sondern mechanistische Unsicherheiten für die Einstufung im Vordergrund standen (z. B. möglicher Schwellenmechanismus, fragliche Gentoxizität bei sonst eindeutiger Befundlage zum kanzerogenen Geschehen).
(3) Kanzerogene, die nach dem nationalen Bewertungsvorschlag der MAK-Kommission in Kategorie 4 oder 5 eingestuft wurden (DFG, 2007), sind in der Regel einer quantitativen Risikoabschätzung zugänglich.
7.2 Information zur Kanzerogenität bei inhalativer Exposition
Turmorgenitätsdaten zum Inhalationspfad sind für eine Ableitung einer Expositions-Risiko-Beziehung am Arbeitsplatz erforderlich oder müssen über Pfad-zu-Pfad-Extrapolationen abschätzbar sein (siehe Nummer 4.3). Liegen Krebsinzidenzen z. B. nur nach oraler, dermaler oder parenteraler Applikation vor, ohne dass eine qualifizierte Pfad-zu-Pfad-Extrapolation möglich wäre, kann keine entsprechende Quantifizierung erfolgen.
7.3 Tumorlokalisationen ohne quantitative Übertragbarkeit
Soweit bestimmte Tumorlokalisationen bei bestimmten Tierspezies auftreten (ggf. auch geschlechtsgebunden oder in Verbindung anderer Stoffeigenschaften) gelten diese Befunde als nicht oder nicht quantitativ übertragbar. Die entsprechenden Einschränkungen sind bei der Prüfung der Minimalkriterien zu beachten (siehe Nummer 4.1).
7.4 Fehlende Studien
Liegen keine tierexperimentellen Langzeitstudien und keine qualifizierten Humanstudien zu einer Substanz vor, ist in der Regel keine Quantifizierung des nominellen Krebsrisikos möglich. In Einzelfällen kann aufgrund von Analogiebetrachtungen und eingeschränkten substanzspezifischen Studien eine Quantifizierung gerechtfertigt sein. Zu den für eine Abschätzung regelmäßig erforderlichen Studien gehören Nachweise für eine mit der Vergleichssubstanz vergleichbaren Gentoxizität. Hierzu sind entsprechende Begründungen vorzulegen.
7.5 Qualität der Studie und der Berichterstattung
(1) Es wird in der Regel eine Veröffentlichung mit detaillierter Berichterstattung vorausgesetzt. Mindestens sollten genannt sein: Spezies, Stamm und Geschlecht der exponierten Tiere und der Kontrolle, Anzahl der exponierten Tiere/ Expositionsgruppe/Geschlecht inkl. Kontrolle, Dosierungen bzw. Luftkonzentration und analytische Nachweismethode für die Expositionsangabe, Gewicht der Tiere zu Beginn und am Ende der Exposition/Vergleich zwischen Expositionsgruppen und Kontrolle, Expositionsdauer und Nachbeobachtungsdauer, Tumorinzidenzen/Gruppe inkl. Kontrolle, Nachweismethode und Untersuchungsumfang zur Feststellung der Tumorinzidenzen, Mortalität während des Versuchs und bei Versuchsende, begleitende nichtbösartige Effekte (Kontrolle, Dosisgruppen) inkl. expositionsbedingten und nichtexpositionsbedingten Effekten, Veränderung der Organgewichte (relativ und absolut), Besonderheiten in der Nahrungszusammensetzung und in der Nahrungsaufnahme, Identität der Substanz inkl. Angabe zur Reinheit bzw. Verunreinigungen, Additive.
(2) Es sollte die Körpergewichtsentwicklung nicht um zehn Prozent oder mehr reduziert sein und die Lebenserwartung der Tiere sollte nicht durch andere Ursachen als Tumorbildung deutlich verringert sein, d. h. die Maximal Tolerierbare Dosis (MTD) sollte nicht überschritten sein.
(3) Werden diese Qualitätskriterien in der Studie oder in der Berichterstattung deutlich unterschritten, so ist bei Einzelfallabwägung in der Regel keine quantitative Abschätzung des Lebenszeitrisikos möglich.
In Versuchsgruppen mit stark erhöhter Tumorhäufigkeit ist auch mit sonstiger Substanztoxizität zu rechnen. Diese steht in der Regel einer Einbeziehung der Gruppe in die Analyse der Expositions-Risiko-Beziehung nicht entgegen.
7.6 Mindestkriterien zur Berücksichtigung von epidemiologischen Studien bei der Risikoableitung
(1) Generelle Anforderungen an epidemiologische Studien: Erfüllen vorhandene epidemiologische Studien zuvor festgelegte Mindestkriterien nicht, so sollten sie bei der Ableitung von Expositions-Risiko-Beziehungen und Arbeitsplatzgrenzwerten nicht berücksichtigt werden. Abweichungen von dieser Regel können mit Begründung erfolgen. Einige der zentralen Anforderungen an epidemiologischen Studien sind:
- 1.
vor Studienbeginn formulierte Studienhypothese/Fragestellung,
- 2.
der Fragestellung/dem nachzuweisenden Risiko angemessener Studienumfang (statistische Power),
- 3.
Berücksichtigung von Confoundern,
- 4.
Vermeidung von Selektionseffekten (Bias) bzw. kritische Diskussion möglicher Auswirkungen auf Studienergebnisse.
- 5.
Informationen, die eine kritische Bewertung der Studienergebnisse erlauben (Berücksichtigung von Konsistenz von Dosis-Wirkungsbeziehungen; Robustheit der Ergebnisse [Sensitivitätsanalysen, z. B. nach Ausschluss bestimmter Subgruppen, stratifiziert nach Beschäftigungsdauer, stratifiziert nach Expositionsintensität etc.])
Aufgrund der kollektiven Expositionszuschreibung sollten Korrelationsstudien für eine Bewertung a priori nicht berücksichtigt werden. Auch Fallstudien ohne Vergleichsgruppe sind für eine Risikoableitung ungeeignet. Querschnittsstudien sind aufgrund des fehlenden Zeitbezugs nur zur Bewertung akuter Effekte geeignet (Monitoring-Studien mit individueller Expositionseinstufung). Ohne Bezug auf einen relevanten Endpunkt sind Querschnittsstudien für die Bewertung eines Krankheitsrisikos nicht geeignet.
Die Deutsche Gesellschaft für Epidemiologie hat Leitlinien für Gute Epidemiologische Praxis (GEP) entwickelt, die dazu dienen, einen Qualitätsstandard für die epidemiologische Forschung in Deutschland zu etablieren und zu helfen, Unredlichkeit und wissenschaftliche Fälschung zu vermeiden und einen vertrauensvollen Umgang unter Wissenschaftlern zu gewährleisten (http://www.dgepi.de/infoboard/stellungnahmen.htm). Die genannten zentralen Anforderungen an epidemiologische Studien sind diesen Leitlinien entnommen.
(2) Die Ermittlung und Bewertung von Expositionen ("exposure assessment") sollte folgende Elemente beinhalten:
- 1.
Darstellung der Methode und der Datenquellen zum "exposure assessment",
- 2.
vorab formulierte Bewertungsregeln für die Expositionsermittlung,
- 3.
Angabe bzw. zumindest Möglichkeit der Berechnung von kumulativen Expositionen, d. h. Angabe der Dauer und Intensität der Exposition,
- 4.
Berücksichtigung von Ko-Expositionen: Im Unterschied zum Experiment treten oft Mischexpositionen auf, die die Zuordnung des Erkrankungsrisikos zu einem speziellen Agens erschweren. Daher müssen in Frage kommende Ko-Expositionen in Betracht gezogen werden.
Sofern diese Elemente nicht hinreichend berücksichtigt sind, erfüllt die Expositionsabschätzung nicht die erforderlichen Mindestkriterien für die Anwendung von Humandaten für die Risikoquantifizierung.
Literatur:
Cordier und Stewart (2005); Ahrens und Stewart (2003); Kromhout (1994)
Die Ermittlung und Bewertung von beruflichen Expositionen ("exposure assessment") erfolgt insbesondere in der Krebsepidemiologie oftmals retrospektiv mit der Gefahr einer Fehlklassifikation der Exposition. Verschiedene Methoden zum "exposure assessment" wurden entwickelt, um eine möglichst valide Einschätzung der beruflichen Expositionen zu ermöglichen. Unabhängig von möglichen Kombinationen und weiteren Informationsquellen beruhen Expositionsermittlungen und -bewertungen im Rahmen arbeitsplatzepidemiologischer Studien auf Messdaten, Experteneinschätzungen, Expositionseinsstufungen durch Job-Expositions-Matrizen (JEM) oder Selbstangaben der Studienteilnehmer/innen. Alle Methoden des "exposure assessments" weisen spezifische Stärken und Schwächen auf. Bei der Ableitung von Expositions-Risiko-Beziehungen können davon unabhängig grundsätzlich alle Methoden berücksichtigt werden, sofern sie die Einschätzung der kumulativen Exposition erlauben.
Weitere Details zu den besonderen Stärken und Schwächen der Studiendesigns: siehe Ahrens et al. (2008).
8 Anforderungen an Dokumentation
8.1 Begründungspapiere
(1) Bei Verwendung im regulatorischen Bereich (z.B. bei Grenzwerten und mit Risikohöhen verknüpften Auflagen an das Risikomanagement) erfordert die Ableitung von stoffbezogenen Expositions-Risiko-Beziehungen und Risikozahlen eine schriftliche, öffentlich zugängliche Begründung (Begründungspapier).
(2) Begründungspapiere können in ihrem methodischen Bezug auf diesen Leitfaden verweisen, so dass z. B. Default-Faktoren oder methodische Einzelschritte bei Übereinstimmung mit dem Leitfaden nicht gesondert im Einzelfall begründet werden müssen. Der Verweis sollte jedoch explizit erfolgen (z. B. "Die Verkürzung der Expositionsdauer wurde entsprechend den Regeln des Leitfadens, Nummer 4.4, berücksichtigt").
(3) Soweit Begründungspapiere auf veröffentlichten Daten beruhen und in der zitierten Quelle alle erforderlichen Angaben enthalten sind (vgl. auch Minimalkriterien nach Nummer 7), ist die eindeutige Zitierung der Quelle zur Beschreibung der Datenbasis einer Risikoquantifizierung ausreichend.
(4) Schwerpunkte eines Begründungspapiers sind
- 1.
Begründungen zum angenommenen vorherrschenden Wirkprinzip (siehe Nummer 2),
- 2.
Abweichungen vom in diesem Leitfaden vorgesehen Default-Vorgehen,
- 3.
Auswahl der Tumorlokalisation (einschließlich Spezies, Geschlecht etc., siehe Nummer 3.1),
- 4.
Darstellung der eigentlichen mathematischen Berechnung. Ferner ergibt sich immer dann ein Begründungsbedarf, wenn dies in einzelnen Nummern dieses Leitfadens explizit gefordert wird.
(5) Ein Verweis auf Risikoquantifizierungen durch Dritte und die dort erfolgten Begründung ist nur dann ausreichend, wenn die zitierte Referenz den Anforderungen dieses Leitfadens in der gewählten Methodik und in der geforderten Transparenz entspricht.
Literatur
- [1]
Ahlbom, A.; Axelson, O.; Hansen, E.S.; Hogstedt, G; Jensen, U.J.; Olsen, J., 1990 Interpretation of "negative" studies in occupational epidemiology Scandinavian Journal of Work, Environment and Health, 16, 1990, 153-157
- [2]
Ahrens, W; Stewart, P., 2003 Retrospective exposure assessment In: Nieuwenhuijsen, M.J., Exposure Assessment in Occupational and Environmental Epidemiology Oxford; New York: Oxford University Press, 2003
- [3]
Ahrens, W.; Behrens, T.; Mester, B.; Schmeißer, N., 2008 Epidemiologie der Arbeitswelt Bundesgesundheitsblatt (im Druck)
- [4]
Attfield, M.D.; Costello, J., 2004 Quantitative exposure-response for silica dust and lung Cancer in Vermont granite workers American Journal of Industrial Medicine, 45, 2004,129-138
- [5]
Becher, H.; Steindorf, K., 1993 Epidemiologische Methoden und Wege der Risikoabschätzung Informatik, Biometrie und Epidemiologie in Medizin und Biologie, 24,1993,14-27
- [6]
Blair, A.; Burg, J.; Foran, J.; Gibb, H.; Greenland, S.; Morris, R.; Raabe, G.; Savitz, D.; Teta, J.; Warteberg, D.; et al., 1995 Guidelines for application of meta-analysis in environmental epidemiology ILSI Risk Science Institute Regulatory Toxicology and Pharmacology, 22, 1995,189-197
- [7]
Bolt, H.; Huici-Montagud, A., 2007 Strategy of the scientific committee on occupational exposure limits (SCOEL) in the derivation of occupational exposure limits for carcinogens and mutagens, Archives of Toxicology, Online-Veröffentlichung, Meeting Reports, November 2007
- [8]
Boobis, A.; Cohen,S.; Dellarco, V; Meek, M.E.; Vickers, C.; Willcock, D.; Farland, W., 2006 IPCS Framework for analyzing the relevance of a cancer mode of action for humans Critical Reviews in Toxicology, 36 (10), 781-792
- [9]
BAuA, Bundesanstalt für Arbeitsschutz und Arbeitsmedizin, 2005 Toleranzschwelle und Akzeptanzschwelle für Gesundheitsrisiken am Arbeitsplatz, 2004-2005, Vorhaben-Nr. F 2010 und F 2080, Auftragsnehmer: FoBiG GmbH, Kalberlah, F., Dortmund, 2005
- [10]
Bundesministerium für Arbeit und Soziales, 2005 Verordnung zum Schutz vor Gefahren (Gefahrstoffverordnung - GefStoffV); 2005 http://www.bmas.de/coremedia/generator/13248/gefstoffv.html
- [11]
Butterworth, B.E., 2006 A classification framework and practical guidance for establishing a mode of action for chemical carcinogens Regulatory Toxicology and Pharmacology, 45, 2006, 9-23
- [12]
Chen, J.; Kodell, R.L.; Gaylor, D.W., 1988 Using the Biological Two-Stage Model to Assess Risk from Short-Term Exposures Risk Analysis, 8 (2), 1988, 223-230
- [13]
Cohen, S.M.; Meek, M.E.; Klaunig, J.E.; Patton, D.E.; Fenner-Crisp, P.A., 2003 The human relevance of information on carcinogenic modes of action: overview. Special issue: Cancer modes of action and human relevance Critical Reviews in Toxicology, 33, 2003, 581-589
- [14]
Cordier, S.; Stewart, P., 2005 Exposure Assessment In: Ahrens, W; Pigeot, I., Handbook of Epidemiology Berlin; Heidelberg; New York: Springer, 2005
- [15]
Crump, K.S.; Howe, R.B., 1984 The multistage model with a time-dependent dose pattern: application to carcinogenic risk assessment Risk Analysis, 4, 1984, 163-176
- [16]
Derelanko und Hollinger, 2002 Handbook of Toxicology, CRC Press, Boca Raton, FL, 2002
- [17]
DFG, Deutsche Forschungsgemeinschaft, 2007 MAK- und BAT-Werte-Liste 2007. Senatskommission zur Prüfung gesundheitsschädlicher Arbeitsstoffe. Mitteilung 43 WILEY-VCH Verlag GmbH, Weinheim, 2007
- [18]
DGEpi, Arbeitsgruppe Epidemiologische Methoden der Deutschen Gesellschaft für Epidemiologie, 2004 Leitlinien und Empfehlungen zur Sicherung von Guter Epidemiologischer Praxis (GEP) Download unter: http://www.dgepi.de/infoboard/stellungnahmen.htm
- [19]
Doll, R.; Wald, N.J., 1994 Interpretation of negative epidemiological evidence for carcinogenicity IARC Scientific Publications Vol. 65 IARC, International Agency for Research on Cancer, Lyon, 1994
- [20]
Dybing, E.; Sanner, T.; Roelfzema, H.; Kroese, D.; Tennant, R.W., 1997 T25: a simplified carcinogenic potency index: description of the System and study of correlations between carcinogenic potency and species/ site specificity and mutagenicity Pharmacology & Toxicology, 80, 1997, 272-279
- [21]
EC, European Commission, 1999 Guidelines for setting specific concentration limits for carcinogens in Annex 1 of Directive 67/548/EEC. Inclusion of potency considerations European Commission, Brüssels, 1999
- [22]
EC, European Commission, 2006 Technical Guidance Document in Support of the Commission Directive 93/ 67/EEC on Risk Assessment for New Notified Substances and the Commission Regulation (EC) 1488/94 on Risk Assessment for Existing Substances and Directive 98/8/EC of the European Parliament and of the Council Concerning the Placing of Biocidal Products on the Market. Human Health Risk Characterisation, Revised Chapter, Final Draft Joint Research Centre, Institute for Health and Consumer Protection, European Chemicals Bureau, Ispra, Italy 2006
- [23]
EFSA, European Food Safety Authority, 2005 Opinion of the Scientific Committee on a request from EFSA related to a harmonised approach for risk assessment of substances which are both genotoxic and carcinogenic (Request No EFSA-Q-2004-020) (Adopted on 18 October 2005) The EFSA Journal, 282, 2005,1-31
- [24]
EPA, Environmental Protection Agency, 2000 Benchmark Dose Technical Guidance Document. Draft U.S. Environmental Protection Agency, Washington, DC, 2000
- [25]
EPA, Environmental Protection Agency, 2005 Guidelines for Carcinogen Risk Assessment. EPA/630/P-03/001F Risk Assessment Forum. U.S. Environmental Protection Agency, Washington, DC, 2005
- [26]
EU, Europäische Union, 2004 Richtlinie 2004/37/EG über den Schutz der Arbeitnehmer gegen Gefährdung durch Karzinogene oder Mutagene bei der Arbeit (Krebsrichtlinie) 2004 http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2004:229:0023:0034:DE:PDF
- [27]
EU, Europäische Union, 2006 Verordnung (EG) Nr. 1907/2006 der Europäischen Parlaments und des Rates vom 18. Dezember 2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH), zur Schaffung einer Europäischen Agentur für chemische Stoffe, zur Änderung der Richtlinie 1999/45/EG und zur Aufhebung der Verordnung (EWG) Nr. 793/93 der Rates, der Verordnung (EG) Nr. 1488/94 der Kommission, der Richtlinie 76/ 769/EWG der Rates sowie der Richtlinien 91/155/ EWG, 93/67/EWG, 93/105/EG und 2000/21/EG der Kommission; 2006 http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2006:396:0001:0851:DE:PDF
- [28]
EU, Europäische Union, 2007 Richtlinie 67/548/ EWG, Anhang VI - Allgemeine Anforderungen für die Einstufung und Kennzeichnung gefährlicher Stoffe und Zubereitungen, letzte Änderungsrichtlinie 2007 http://www.baua.de/de/Themen-von-A-Z/Gefahrstoffe/Rechtstexte/EG-Richtlinien.html__nnn=true
- [29]
FDA, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), 2001 Guidance for Industry. Statistical Aspects of the Design, Analysis, and Interpretation of Chronic Rodent Carcinogenicity Studies of Pharmaceuticals http://www.fda.gov/cder/guidance/index.htm
- [30]
Foth, H.; Degen, G.H.; Bolt, H.M., 2005 New aspects in the Classification of carcinogens Arhiv za Higijenu Rada i Toksikologiju, 56, 2005, 167-175
- [31]
Goldbohm, R.A.; Tielemans, E.L.J.P.; Heederik, D.; Rubingh, CM.; Dekkers, S.; Willems, M.I.; Kroese, E.D., 2006 Risk estimation for carcinogens based on epidemiological data: A structured approach, illustrated by an example on chromium Regulatory Toxicology and Pharmacology, 44, 2006, 294-310
- [32]
HEI-AR, Health Effects Institute - Asbestos Research, 1991 Asbestos in public and commercial buildings: A literature review and synthesis of current knowledge Health Effects Institute - Asbestos Research, Cambridge, MA, 1991
- [33]
Hengstler, J.G.; Degen, G.H.; Foth, H.; Bolt, H.M., 2006 Thresholds for specific classes of genotoxic carcinogens: a new strategy for carcinogenicity categorisation of chemicals to be published in SIIC, in press, 2006
- [34]
Kalberlah, F.; Schneider, K., 1998 Quantifizierung von Extrapolationsfaktoren. Endbericht des Forschungsvorhabens Nr. 116 06 113 des Umweltbundesamtes Schriftenreihe der Bundesanstalt für Arbeitsschutz und Arbeitsmedizin, Dortmund, Fb 796 Wirtschaftsverlag NW, Bremerhaven, 1998
- [35]
Kirkland, DJ.; Aardema, M.; Banduhn, N.; Carmichael, P.; Fautz, R.; Meunier, J.-R.; Pfuhler, S., 2007a In vitro approaches to develop weight of evidence (WoE) and mode of action (MoA) discussions with positive in vitro genotoxicity results Mutagenesis, 22, 2007, 161 - 175
- [36]
Kirkland, D.; Pfuhler, S.; Tweats, D.; Aardema, M.; Corvi, R.; Darroudi, F.; Elhajouji, A.; Glatt, H.; Hastwell, P.; Hayashi, M.; Kasper, P.; Kirchner, S.; Lynch, A.; Marzin, D.; Maurici, D.; Meunier, J.R.; Müller, L.; Nohynek, G.; Parry, J.; Parry, E.; Thybaud, V.; Tice, R.; van Benthem, J.; Vanparys, P.; White, P., 2007b How to reduce false positive results when undertaking in vitro genotoxicity testing and thus avoid unnecessary follow-up animal tests: Report of an ECVAM Workshop Mutation Research, 628, 2007, 31-55
- [37]
Kirman, CR.; Sweeny, L.M.; Teta, M.J.; Sielken, R.L.; Valdez-Flores, C.; Albertini, R.J.; Gargas, M.L., 2004 Addressing nonlinearity in the exposure-response relationship for a genotoxic carcinogen: Cancer potency estimates for ethylene oxide Risk Analysis, 24, 2004, 1165-1183
- [38]
Kromhout, H., 1994 From eyeballing to Statistical modelling: methods for assessment of occupational exposure PhD Thesis, Agricultural University of Wageningen, 1994
- [39]
Last J.M, 2001 A dictionary of epidemiology, 4th ed. Oxford: Oxford University Press, 2001
- [40]
Lutz, W.K., 2000 A true threshold dose in chemical carcinogenesis cannot be defined for a population, irre-spective of the mode of action Human & Experimental Toxicology, 19 (10), 2000, 566-568; discussion 571-572
- [41]
McConnell, E.E.; Swenberg, J.A., 1994 Review of styrene and styrene oxide long-term animal studies Critical Reviews in Toxicology, 24, Suppl., 1994, 49-55
- [42]
McConnell, E.E.; Solleveld, H.A.; Swenberg, J.A.; Boorman, G.A., 1986 Guidelines for combining neoplasms for evaluation of rodent carcinogenesis studies Journal of the National Cancer Institute, 76,1986,283-289
- [43]
Meek, M.E.; Bucher, J.R.; Cohen, S.M.; Dellarco, V.; Hill, R.N.; Lehman-McKeeman, L.D.; Longfellow, D.G.; Pastoor, T.; Seed, J.; Patton, D.E., 2003 A framework for human relevance analysis of Information on carcinogenic modes of action: special issue: Cancer modes of action and human relevance Critical Reviews in Toxicology, 33, 2003, 591-653
- [44]
Neumann, H.-G., 2006a Die Risikobewertung von Kanzerogenen und die Wirkungsschwelle, Teil I Bundesgesundheitsblatt, 49, 2006, 665-674
- [45]
Neumann, H.-G., 2006b Die Risikobewertung von Kanzerogenen und die Wirkungsschwelle, Teil II Bundesgesundheitsblatt, 49, 2006, 818-823
- [46]
Neumann, H.-G., 2006c Die Risikobewertung von Kanzerogenen und die Wirkungsschwelle, Teil III Bundesgesundheitsblatt, 49, 2006, 911-920
- [47]
o.V., 1998 Kriterien für die Ableitung von gesundheitsbasierten Luftgrenzwerten bei limitierter Datenlage Bundesarbeitsblatt, 10,1998, 74-76
- [48]
Preston, R.J.; Williams, G.M., 2005 DNA-reactive carcinogens: mode of action and human Cancer hazard Critical Reviews in Toxicology, 35, 2005, 673-683
- [49]
Rice, EL.; Park, R.; Stayner, L.; Smith, R.; Gilbert, S.; Checkoway, H., 2001 Crystalline silica exposure and lung cancer mortality in diatomaceous earth industry workers: a quantitative risk assessment Occupational and Environmental Medicine, 58, 2001, 38-45
- [50]
Roller, M., 2005 Persönliche Mitteilung, Arbeitskreis CMR des Unterausschuss III, Ausschuss für Gefahrstoffe, März 2005
- [51]
Roller, M.; Akkan, Z.; Hassauer, M.; Kalberlah, F., 2006 Risikoextrapolation vom Versuchstier auf den Menschen bei Kanzerogenen Fb 1078, Bundesanstalt für Arbeitsschutz und Arbeitsmedizin, Dortmund/Berlin/ Dresden Wirtschaftsverlag NW, Bremerhaven, 2006
- [52]
Sanner, T.; Dybing, E.; Willems, M.I.; Kroese, E.D., 2001 Arsimple method for quantitative risk assessment of non-threshold carcinogens based on the dose descriptor T25 Pharmacology & Toxicology, 88, 2001, 331-341
- [53]
SCOEL, Scientific Committee on Occupational Exposure Limits, 2003 Recommendation from the Scientific Committee on Occupational Exposure Limits: risk assessment for 1,3-butadiene SCOEL/SUM/75 final. November 2003
- [54]
Seed, J.; Carney, E.W.; Corley, R.A.; Crofton, K.M.; DeSesso, J.M.; Foster, P.M.D.; Kavlock, R.; Kimmel, G.; Klaunig, J.; Meek, M.E.; Preston, R.J.; Slikker, W; Tabacova, S.; Williams, G.M.; Wiltse, J.; Zoeller, R.T.; Fenner-Crisp, P; Patton, D.E., 2005 Overview: Using mode of action and life stage information to evaluate the human relevance of animal toxicity data Critical Reviews in Toxicology, 35, 2005, 664-672
- [55]
Sorahan, T; Hamilton, L.; Gompertz, D.; Levy, L.S.; Harrington, J.M., 1998 Quantitative risk assessments derived from occupational Cancer epidemiology: a worked example Annais of Occupational Hygiene, 42, 1998, 347-352
- [56]
Statistisches Bundesamt Deutschland, 2006
- [57]
Statistisches Jahrbuch für die Bundesrepublik Deutschland 2006 https://www-ec.destatis.de/csp/shop/sfg/bpm.html.cms.cBroker.cls?cmspath=struktur,vollanzeige.csp&ID=1019209
- [58]
Stayner, L.; Smith, R.; Bailer, A.J.; Luebeck, E.G.; Moolgavkar, S.H., 1995 Modeling epidemiologic studies of occupational cohorts for the quantitative assessment of carcinogenic hazards American Journal of Industrial Medicine, 27,1995, 155-170
- [59]
Stayner, L.; Dankovic, D.; Smith, R.; Steenland, K., 1998 Predicted lung cancer risk among miners exposed to diesel exhaust particles American Journal of Industrial Medicine, 34,1998, 207-219
- [60]
Stayner, L.T.; Dankovic, D.A.; Smith, R.J.; Gilbert, S.J.; Bailer, A.J., 2000 Human cancer risk and exposure to 1,3-butadiene - a tale of mice and men Scandinavian Journal of Work, Environment and Health, 26, 2000, 322-330
- [61]
Steenland, K.; Mannetje, A.; Boffetta, P; Stayner, L.; Attfield, M.; Chen, J.; Dosemeci, M.; DeKlerk, N.; Hnizdo, E.; Koskela, R.-S.; Checkoway, H., 2001 Pooled exposure-response analyses and risk assessment for lung cancer in 10 cohorts of silica-exposed workers: an IARC multicentre study Cancer Causes & Control, 12, 2001, 773-784
- [62]
Streffer, C.; Bolt, H.M.; Follesdal, D.; Hall, P; Hengstler, J.G.; Jacob, P; Oughton, D.; Priess, K.; Rehbinder, E.; Swaton, E., 2004 Environmental Standards - dose-effect relations in the low dose ränge and risk evaluation Springer Verlag Berlin, 2004
- [63]
Umweltbundesamt, 2003 Vergleich der Verfahren zur Ableitung gesundheitsbezogener Wirkungsschwellen (Benchmark - NOAEL). Abschlussbericht. Forschungs- und Entwicklungsvorhaben FKZ 201 65 201/01 (Autoren: Kalberlah, F. und Hassauer, M.) Freiburg/ Berlin, 2003 http://www.apug.de/Geschaeftsstelle/Liste/Benchmark_Endbericht.pdf
- [64]
Van Wijngaarden, E.; Hertz-Picciotto, I., 2004 A simple approach to performing quantitative cancer risk assessment using published results from occupational epidemiology studies The Science of the Total Environment, 332, 2004, 81-87
- [65]
WHO, World Health Organization, o.J. The IPCS Conceptual Framework for Cancer Risk Assessment http://www.who.int/ipcs/methods/harmonization/en/MOA_text.pdf
- [66]
Yamasaki, H., 1988 Multistage carcinogenesis: implications for risk estimation Cancer and Metastasis Reviews, 7, 1988, 5-18
10 ANHÄNGE
10.1 Glossar
Additional Risk:
Berechnungsweise des expositionsbedingten Lebenszeitrisikos als Differenz zwischen dem Risiko der Exponierten und dem Risiko der nicht-exponierten Kontrollgruppe:
PA(x) = P(x)-P(0)
| mit | PA(x): | "additional risk" bei der Exposition x |
|---|---|---|
| P(x): | Lebenszeitrisiko der Exponierten | |
| P(0): | "Hintergrundrisiko" (Lebenszeitrisiko einer nicht-exponierten Kontrollgruppe) |
Der Begriff des "additional risk" wird insbesondere für Daten aus Tierversuchen verwendet, im Falle epidemiologischer Daten wird für das analoge Risiko eher der Begriff des Exzess-Risikos (siehe dort) verwendet.
Adduktbildung:
Hier: Bindung eines Fremdstoffs oder dessen Stoffwechselprodukt an die DNA. DNA-Addukte im Zellkern können unter Umständen die Zellteilung verhindern oder Mutationen auslösen.
AIC (Akaike Information Criterion):
Statistisches Verfahren zur Beschreibung der relativen Anpassungsgüte von Kurvenmodellierungen. In der Regel ergeben besser angepasste Kurven niedrigere AIC-Werte. Wichtiger Test im Benchmarkdose-Verfahren (siehe dort).
Akzeptables/tolerables Risiko:
Beim gesundheitlichen Risiko (siehe dort) durch Einwirkung von Gefahrstoffen handelt sich um ein Kontinuum, das nach einem vom Ausschuss für Gefahrstoffe (AGS) übernommenen Ansatz durch zwei Zäsurpunkte in folgende drei Bereiche unterteilt werden kann:
Ist ein Schadenseintritt lediglich möglich, wird das damit verbundene Risiko als "hinnehmbar" (akzeptabel) bewertet. Bei diesem Risiko sind die Grundmaßnahmen zum Schutz der Beschäftigten erforderlich (Bereich unterhalb des Akzeptanzrisikos).
Ist ein Schadenseintritt nicht bereits hinreichend wahrscheinlich und nicht lediglich möglich, wird das damit verbundene Risiko als "unerwünscht" bewertet. Dieses Risiko bezeichnet die Besorgnis eines Gesundheitsschadens.
Ist ein Schadenseintritt hinreichend wahrscheinlich, wird das damit verbundene Risiko als "nicht hinnehmbar" bewertet. Dieses Risiko bezeichnet eine Gefahr für die Gesundheit (oberhalb des Toleranzrisikos).
Die Risikohöhen für die bezeichneten Zäsurpunkte (Akzeptanz-, Toleranzrisiko) können nicht wissenschaftlich begründet, sondern nur gesellschaftspolitisch gesetzt werden. Dabei sind eine Reihe von Kriterien zu beachten, neben der Risikowahrnehmung sind dies z. B. die Schwere eines Gesundheitsschadens, das mögliche Schadensausmaß (Art des Schadens und/oder die Anzahl der Betroffenen), die Relation zu vergleichbaren anderen Arbeitsplatzrisiken, ein unmittelbarer Nutzen und die tatsächlichen und möglichen Risikominderungsmaßnahmen.
Allometrisches Scaling:
Element der Interspeziesextrapolation (siehe dort) von kleinen Versuchstieren auf den Menschen. Unter Allometrie versteht man die Ermittlung der Abhängigkeit verschiedener biologischer Parameter von der Körpergröße. Beim allometrischen Scaling wird rechnerisch berücksichtigt, dass z. B. bei Säugern die Stoffwechselaktivität nicht linear mit dem Körpergewicht der verschiedenen Tierarten ansteigt. Dies hat zur Folge, dass der Mensch gegen vergleichbare toxische Einflüsse empfindlicher zu sein scheint als beispielsweise die Maus, wenn man die aufgenommene Dosis auf das Körpergewicht bezieht.
Alpha-2u-globulin:
Niedermolekularer Eiweißstoff, der in großen Mengen in der Leber erwachsener männlicher Ratten gebildet wird. Bestimmte leichte Kohlenwasserstoffe (z. B. Isophoron, 1,4-Dichlorbenzol, Limonen) binden an Alpha-2u-globulin. Die so entstehenden Komplexe reichern sich in den Nierenzellen an, was in der Folge zu Zelluntergang mit anschließender Reparatur, Regeneration und vermehrtem Auftreten von Nierentumoren führen kann. Dieser nichtgentoxische Tumorentstehungsmechanismus (siehe "Gentoxizität") wird als geschlechts- und speziesspezifisch und ohne Relevanz für den Menschen angesehen (siehe auch Nummer 10.3).
Aneuploidie:
Abweichung von der Zahl des normalen (euploiden) Chromosomensatzes um ein oder mehrere Chromosomen.
Attributables Risiko:
Das Attributable Risiko oder Attributivrisiko bezeichnet den Anteil der Krankheitsbelastung in der Bevölkerung, der auf einen bestimmten Risikofaktor (siehe dort) zurückzuführen ist. Zur Berechnung des Attributivrisikos sind zwei Informationen erforderlich:
die Häufigkeit des Risikofaktors in der Bevölkerung,
das Ausmaß der Erhöhung des Erkrankungsrisikos durch diesen Risikofaktor.
Nimmt man beispielsweise an, dass bei starken Rauchern das Risiko, an Lungenkrebs zu erkranken im Vergleich zu Nicht-Rauchern auf das Zehnfache ansteigt, und nimmt man weiter an, dass die Häufigkeit des Rauchens bei Männern in der Bevölkerung 40 Prozent beträgt, dann würde sich das Attributiv-Risiko auf etwa 78 Prozent belaufen. In vergleichbarer Weise lässt sich für berufliche Expositionen auf der Basis von Daten zur Expositions-Prävalenz und unter Verwendung von Risiko-Schätzungen aus vorliegenden Studien zur jeweiligen Exposition schätzen.
Man unterscheidet das Attributable Risiko unter Exponierten (ARE) vom Attributablen Risiko in der Allgemeinbevölkerung (PAR). Während das ARE angibt, welcher Anteil der Erkrankungsfälle in der exponierten Teilbevölkerung auf die Exposition zurückzuführen ist, gibt das PAR den entsprechenden Anteil für die Gesamtbevölkerung an. Bei seltenen Expositionen kann daher das PAR zwar klein sein, jedoch kann bei entsprechender Höhe des Relativen Risikos (RR) der diesbezügliche Anteil in der exponierten Teilgruppen, wie z. B. den Beschäftigten in einem bestimmten Produktionszweig, sehr hoch sein, und z. B. bei einem RR > 2 über 50 Prozent liegen.
Mathematische Definitionen von ARE und PAR:

Benchmark-Verfahren:
Anpassung eines mathematischen Modells an die in einer Studie erhobenen Daten zum Dosis-Wirkungs-Zusammenhang. Dafür stehen mehrere Modellfunktionen zur Verfügung.
Das Benchmark-Verfahren ist ein Instrument zur Ermittlung eines "point of departure" (siehe dort) für quantitative Risikoabschätzungen. Für eine definierte Effekthäufigkeit bzw. ein definiertes Effektmaß, der sog. "benchmark response" (BMR), kann die Dosis geschätzt werden, die mit einer bestimmten Wahrscheinlichkeit zu diesem Effekt führt. Diese Dosis nennt man "benchmark dose" (BMD). Eine BMD10 wäre diejenige Dosis, die ein zehnprozentiges Risiko für das Auftreten eines interessierenden Effekts erwarten lässt. Die Sicherheit der Abschätzung einer Dosis-Wirkungs-Beziehung wird durch Angabe eines Vertrauensbereichs quantifiziert. Den Wert des unteren (in der Regel 90- oder 95 Prozent-) Vertrauensbereichs der "benchmark dose" bezeichnet man als "benchmark dose lower bound" (BMDL). Eine Prüfung der Anpassungsgüte der Ergebnisse mit verschiedenen Modellfunktionen kann anhand des AIC erfolgen (siehe dort).
Bias:
Unter dem Begriff Bias versteht man in der Epidemiologie eine Verzerrung, die auf einen systematischen Fehler bei der Erhebung der Daten zurückzuführen ist. Im Gegensatz zu zufälligen Fehlern führen systematische Fehler zu einseitigen Abweichungen.
BMD (Benchmark Dose):
siehe " Benchmark-Verfahren"
BMDL (Benchmark Dose Lower Bound):
siehe "Benchmark-Verfahren"
BMR (Benchmark Response):
siehe " Benchmark-Verfahren"
Chi-Quadrat- Verteilung:
ist eine stetige Wahrscheinlichkeitsverteilung über die Menge der positiven reellen Zahlen
Confounder:
Störvariable, die sowohl mit dem eigentlich untersuchten Einfluss (z. B. bestimmter Arbeitsstoff) als auch mit dem untersuchten Endpunkt (z. B. Krebsentstehung) assoziiert ist. Confounding ist das Vermischen von Störfaktoren-Effekten mit dem Effekt desjenigen Risikofaktors, der untersucht wird.
Default:
Statistisch gestützter Standardwert oder -annähme, der oder die in Ermangelung Stoff- oder Tierart-spezifischer Daten verwendet werden soll. Ein Default lässt Abweichungen zu und ist ein Mittel, um Systeme zu beschreiben, deren Merkmale nicht vollständig bekannt sind.
Dosis-Wirkungs-Beziehung:
Funktionale Beziehung zwischen Dosis und Wirkung (Effektstärke) einer pharmakologisch oder toxikologisch aktiven Substanz. Dosis-Wirkungs-Beziehungen für den Endpunkt Krebs sind streng genommen Dosis-Häufigkeits-Beziehungen und beschreiben die Tumorrate in Abhängigkeit von der Dosis (oder Konzentration). Diese Funktionen sind stetig und nähern sich meist asymptotisch einem maximalen Wert für die Tumorrate.
Für den - tierexperimentell in der Regel nicht zugänglichen - Niedrigdosisbereich sind mehrere Kurvenverläufe, z. B. mit dem Benchmark-Verfahren (siehe dort), modellierbar:
Lineare Dosis-Wirkungs-Beziehung: Kurvenabschnitt lässt sich durch eine Geradenfunktion beschreiben.
Sublineare Dosis-Wirkungs-Beziehung: Der mit wachsender Dosis zunächst langsame Anstieg z. B. der Tumorrate beschleunigt sich überproportional bei zunehmender Dosiserhöhung ("nach unten durchhängende" Kurve).
Supralineare Dosis-Wirkungs-Beziehung: Kleinere Dosiserhöhungen im Niedrigdosisbereich bewirken einen relativ steilen Anstieg z. B. der Tumorrate, während darüber hinausgehende Erhöhungen der Dosis nur noch zu einer geringen Zunahme der Tumorrate und damit zu einer Abflachung der Kurve führen ("nach oben ausgebauchte" Kurve).
Diese Beschreibungen der Kurvenverläufe beinhalten grundsätzliche keine Informationen darüber, ob die Funktionen durch den Nullpunkt verlaufen oder nicht.
EFSA-Konzept:
Strategie der Europäischen Behörde für Lebensmittelsicherheit (EFSA) für die Risikobewertung von gentoxischen (siehe "Gentoxizität") und krebserzeugenden Stoffen. Das Konzept beruht auf der Berechnung eines "margin ofexposure" (siehe dort). Als Bezugspunkt auf der Dosis-Wirkungs-Kurve wird die Dosis ermittelt, die im Tierexperiment eine Tumorrate von zehn Prozent bewirkt (bei befriedigender Datenlage berechnet als BMDL, siehe dort). Liegt der "margin of exposure" (also das Verhältnis zwischen über den Verdauungstrakt aufgenommener Dosis und BMDL10) bei 10.000 oder höher, wird das Krebsrisiko für Konsumenten von belasteten Lebensmitteln als gering eingestuft und vorgeschlagen, diese Substanzen mit niedriger Priorität zu behandeln. Je weiter der "margin of exposure" unter 10.000 liegt, desto dringlicher werden Minimierungsmaßnahmen.
Enzyminduktion:
Steigerung der Synthese von bestimmten Enzymen in den Zellen eines Gewebes. Werden Stoffwechselenzyme induziert, kann dies Auswirkung auf die Entgiftung oder Giftung aufgenommener Fremdstoffe haben.
Epidemiologie:
Epidemiologie ist die Untersuchung der Verteilung und der Ursachen von gesundheitsbezogenen Zuständen oder Ereignissen in definierten Populationen (siehe dort) und die Anwendung der Ergebnisse derartiger Untersuchungen mit dem Ziel, Gesundheitsprobleme zu vermeiden. Unter "Untersuchung" sind Beobachtungsstudien, Surveys, Hypothesentests, analytische und experimentelle Studien zu verstehen. "Verteilung" beinhaltet die Auswertung von entsprechenden Daten nach Zeit, Ort und Personengruppen. Unter "Ursachen" sind alle physikalischen, biologischen, sozialen, kulturellen und verhaltensbedingten Faktoren zu verstehen, die einen Einfluss auf die Gesundheit haben können. "Gesundheitsbezogene Zustände oder Ereignisse" umfassen Erkrankungen, Todesursachen, Verhaltensweisen wie z. B. Tabakkonsum, Reaktionen auf Präventionsmaßnahmen und die Bereitstellung und Nutzung von Gesundheitsdiensten. Unter "definierten Populationen" sind Menschengruppen mit identifizierbaren Charakteristika (Alter, Geschlecht, Wohnort etc.) zu verstehen. Mit "Anwendung der Ergebnisse..." wird explizit auf das Ziel von Epidemiologie, nämlich die Förderung, den Schutz und die Wiederherstellung von Gesundheit, hingewiesen (nach Last, 2001).
Exzess-Risiko:
Mehrdeutige Begriffsverwendung:
- 1.
Oft definiert als das zusätzliche Erkrankungsrisiko unter Exponierten in Relation zum Basisrisiko, auch relative Risikodifferenz (RD) genannt: RD=RR-1. Sie gibt die prozentuale Risikoerhöhung unter Exponierten an. Bei einem Relativen Risiko (RR) von z. B. 1,5 beträgt diese 50 Prozent, bei einem RR von 2,0 100 Prozent und bei einem RR von 10 entsprechend 900 Prozent.
- 2.
In diesem Leitfaden wird darunter das expositionsbedingte Lebenszeitrisiko verstanden, das in der Regel als Differenz zwischen dem Risiko der Exponierten und dem Risiko einer nicht-exponierten Vergleichsgruppe (z. B. der Allgemeinbevölkerung) definiert wird:
Pexcess (x) = P(x) - P(0) mit Pexcess (x): Exzess-Risiko bei der Exposition x P(x): Lebenszeitrisiko der Exponierten P(0): "Hintergrundrisiko" (Lebenszeitrisiko einer nicht-exponierten Vergleichsgruppe)
Der so verstandene Begriff des Exzess-Risikos wird insbesondere bei epidemiologischen Daten verwendet, es ist dabei formal identisch mit dem "additional risk" (siehe dort). Der Begriff des Exzess-Risikos mag im Falle von Tierversuchen - formal nicht ganz korrekt - auch dann angewandt werden, wenn das expositionsbedingte Lebenszeitrisiko als "extra risk" (siehe dort) berechnet wurde.
Extrapolationsfaktor/Sicherheitsfaktor:
Ein Extrapolationsfaktor ist physiologisch/empirisch begründet. Bei der Risikoabschätzung geht man von vorliegenden toxikologischen Daten aus und extrapoliert auf einen experimentell nicht ermittelten Erwartungswert (z. B. Absenkung der Effektkonzentration bei Verlängerung der Versuchsdauer). Diese quantitative Abschätzung muss eine nachvollziehbare Interpretation empirischer Daten einschließen.
Die Berücksichtigung darüber hinaus gehender, eher qualitativer Aspekte (Datengüte, Schwere des Effekts, Verdachtsmomente) erfolgt, um nach dem Vorsorgeprinzip auch vor unbekannten oder wissenschaftlich/empirisch nicht quantifizierbaren Risiken zu schützen. Ein hierfür eingesetzter Faktor wird als Sicherheitsfaktor bezeichnet.
Extra Risk:
Berechnungsweise des expositionsbedingten Lebenszeitrisikos anhand des Risikos der Exponierten und des Risikos einer nicht-exponierten Kontrollgruppe gemäß folgender Formel:
| PE (x) = [P(x)-P(0)] : [1-P(0)] | |
|---|---|
| mit PE (x): | "extra risk" bei der Exposition x |
| P(x): | Lebenszeitrisiko der Exponierten |
| P(0): | "Hintergrundrisiko" (Lebenszeitrisiko einer nicht-exponierten Kontrollgruppe) |
Es handelt sich somit um den Quotienten aus "additional risk" (siehe dort) und dem Anteil der Individuen, die bei Abwesenheit einer Exposition nicht reagieren. Die Berechnungsweise des "extra risk" wird aus rechentechnischen Gründen bei bestimmten Dosis-Wirkungsmodellen insbesondere bei Daten aus Tierversuchen verwendet; das Ergebnis unterscheidet sich in der Regel wenig vom "additional risk".
Fall-Kontroll-Studie:
Das Ziel von Fall-Kontroll-Studien ist es, die Bedeutung von Risikofaktoren (siehe dort) für die Entstehung von Krankheiten quantitativ zu ermitteln. Die logische Basis für Fall-Kontroll-Studien ergibt sich aus der Überlegung, dass ein Risikofaktor, der die Entstehung einer Krankheit begünstigt, bei Patienten mit dieser Krankheit vor Krankheitsbeginn häufiger vorhanden gewesen sein muss als in einer Vergleichsgruppe von Nicht-Erkrankten. Da bei Fall-Kontroll-Studien die Recherchen erst nach eingetretener Erkrankung einsetzen, also in die Vergangenheit gerichtet sind, gehören die Fall-Kontroll-Studien zu den retrospektiven Studienformen. Das Ergebnis einer Fall-Kontroll-Studie ist die sogenannte Odds Ratio (siehe dort), eine Verhältniszahl, die angibt, um wie viele Male häufiger die Erkrankung bei vorhandenem Risikofaktor auftritt als ohne. Eine Odds Ratio unter 1.0 würde ein erniedrigtes Risiko anzeigen, ein Wert über 1.0 ein erhöhtes. Eine Odds Ratio von 1.5 entspricht einer Risiko-Erhöhung um 50 Prozent. Zur Bewertung der Relevanz einer Odds Ratio ist allerdings die Berechnung eines zugehörigen Konfidenz-Intervalls (siehe dort) unerlässlich.
In eine Kohorte eingebettete Fall-Kontroll-Studie: Dieses Design stellt einen Sonderfall der Fall-Kontroll-Studie dar, welches in der Arbeitsepidemiologie häufig vorkommt. Alle Fälle aus einer Kohorte werden mit einem zufälligen Sample der zum Zeitpunkt der Falldiagnose nicht erkrankten Kontrollpersonen aus derselben Kohorte verglichen ('incidence density sampling'), wodurch die optimalen Bedingungen einer inzidenten und vollständigen Fallrekrutierung sowie die zufällige Auswahl nicht erkrankter Personen aus derselben Bezugspopulation erfüllt werden.
First-Pass-Effekt:
Stoffe, die über den Verdauungstrakt aufgenommen werden, gelangen nach Resorption über die Pfortader in die Leber. Bei ihrer ersten Leberpassage ("firstpass") können sie teilweise in erheblichem Umfang verstoffwechselt werden, so dass nur noch ein Bruchteil der Ausgangssubstanz die anderen Organe erreicht.
Gamma-Funktion:
Spezielle mathematische Funktion, aus der sich eine kontinuierliche Wahrscheinlichkeitsverteilung (Gamma-Verteilung) ableitet.
Gavage:
Verabreichung einer Substanz mittels Schlundsonde.
Gentoxisch:
Toxisch für das Genom; schädigende Wirkung auf das genetische Material in Zellen. Ein Oberbegriff der neben der Induktion von Gen-, Chromosomen- oder Genommutationen auch solche Effekte umfasst, die in Indikatortests (z. B. SOS-Chromotest, Comet Assay) nachgewiesen werden. Diese Wirkungen können direkt, durch den Ausgangsstoff oder indirekt, durch Stoffwechselprodukte, ausgelöst werden. Gentoxische Substanzen können Mutationen und Tumoren auslösen.
Man unterscheidet zwischen
primär gentoxisch wirkenden Substanzen: Ausgangsstoff und/oder Stoffwechselprodukt(e) reagieren unmittelbar mit der DNA und können so die genetische Information verändern;
sekundär gentoxisch wirkenden Substanzen: Induktion von genetischen Schäden ohne unmittelbare Wechselwirkung mit der DNA. Beispiele sind oxidative Schädigung durch entstehende reaktive Sauerstoffspezies oder Störung der DNA-Reparatur.
Hardersche Drüse:
Zusätzliche Tränendrüse der Nickhaut im nasenseitigen Augenwinkel von vielen Tierspezies. Der Mensch besitzt keine Nickhaut.
hT25:
Humanäquivalente T25 (siehe dort), die durch die Extrapolation auf den Menschen aus der aus Tierversuchsdaten ermittelten T25 errechnet ist.
Hypophyse:
Hirnanhangsdrüse, die eine Reihe von Hormonen produziert.
Interspeziesextrapolation:
Hier: Umrechnung von tierexperimentell erhaltenen Ergebnissen auf die (durchschnittlichen) Verhältnisse beim Menschen.
Intraspeziesextrapolation:
Hier: rechnerische Berücksichtigung von Empfindlichkeitsunterschieden innerhalb der menschlichen Bevölkerung bei der Risikoabschätzung.
Inzidenz:
Bezeichnet die Häufigkeit der Neuerkrankungen an einer bestimmten Erkrankung bezogen auf einen definierten Zeitraum (meistens ein Jahr) und bezogen auf eine definierte Population. Zur Ermittlung der Inzidenz müssen in einer definierten Region alle neu erkrankten Patienten erfasst werden. Dieses ist in der Regel durch bevölkerungsbezogene epidemiologische Krankheitsregister möglich, z. B. Krebsregister, Herzinfarktregister, oder durch eigene Inzidenz-Studien. Für Deutschland kann die Inzidenz nur für wenige Krankheitsgruppen und regional sehr beschränkt angegeben werden. Das einzige epidemiologische Krebsregister, das für alle Altersgruppen verlässliche Inzidenzdaten über längere Zeiträume liefert, ist das Krebsregister des Saarlandes und das Krebsregister der ehemaligen DDR bis 1990. Die aufgrund des Bundeskrebsregistergesetzes seit den 90er Jahren aufgebauten Landeskrebsregister sind nur teilweise vollständig, werden aber zukünftig vermehrt nutzbare Daten liefern (siehe "Dachdokumentation Krebs" unter www.rki.de). Für Malignome des Kindesalters (bis zum vollendeten 14. Lebensjahr) liefert das Mainzer Kinderkrebsregister Daten für die gesamte Bundesrepublik.
Die kumulative Inzidenz (Cl) gibt den Anteil neu erkrankter Personen zu einem definierten Zeitpunkt für eine spezifische Erkrankung an:

Klitorisdrüse:
siehe "Präputialdrüse"
Kohorten-Studie:
Eine Kohorte bezeichnet in der Epidemiologie eine Gruppe von Personen, die durch ein gemeinsames Merkmal charakterisiert ist. Dieses Merkmal kann eine gemeinsame Exposition gegenüber einem Schadstoff sein, das Wohnen in einer bestimmten Region, der gleiche Beruf oder Ähnliches. In einer Kohorten-Studie werden die Mitglieder der Kohorte über einen definierten Zeitraum beobachtet auf das Auftreten von Endpunkten hin. Diese Endpunkte können das Auftreten definierter Erkrankungen oder das Versterben an definierten Todesursachen sein. Da sie von einer Exposition ausgehend das zeitlich nachfolgende Erkrankungsrisiko untersuchen, handelt es sich um ein prospektives Studiendesign. In der Arbeitsmedizin wird der Ausgangspunkt von Kohortenstudien häufig zeitlich zurückverlegt. Diese Studien werden oft als historische Kohortenstudien oder als historisch-prospektives Design bezeichnet.
Wie auch tierexperimentelle Studien erfordern epidemiologische Studien bei ihrer Planung die Festlegung der erforderlichen Stichprobenumfänge.
Konfidenz-Intervall:
Ein Konfidenz-Intervall erlaubt die Beurteilung der Schwankungsbreite einer Schätzung (z. B. Odds Ratio, Relatives Risiko, Standardisierte Mortalitäts-Ratio). Das Intervall gibt an, in welchen Bereich 95 von 100 möglichen Schätzungen fallen würden (wenn das 95-Prozent-Konfidenz-Intervall berechnet wurde) oder 99 von 100, wenn das 99-Prozent-Konfidenz-Intervall angegeben wurde. Gebräuchliche ist das 95-Prozent-Konfidenz-Intervall. Wenn eine Odds Ratio (siehe dort) mit 1.41 geschätzt wurde und das Konfidenz-Intervall von 0.95 bis 1.67 reicht, ist keine signifikante Erhöhung der Odds Ratio zu konstatieren, weil in den 95-Prozent-Bereich auch Werte unter 1.0 fallen, d. h. erniedrigte Risiken wie Risikoerhöhungen auftreten können.
Korrelationsstudien:
siehe " Ökologische Studien"
Leydigzell-Tumor:
Neubildung, die ihren Ausgang in den Testosteron-produzierenden Leydigzellen des Hodens nimmt. Während Leydigzelltumoren beim Menschen sehr selten auftreten, wird insbesondere beialternden Fischer-344-Laborratten eine hohe spontane Inzidenz beobachtet (siehe auch Nummer 10.3).
Margin of Exposure (MoE):
Abstand zwischen der aus experimentellen Daten abgeleiteten Konzentration, die noch toxische Effekte (hier: Tumoren) auslöst und der erwarteten bzw. durch Messungen ermittelten Konzentration, gegen die der Mensch (am Arbeitsplatz) exponiert ist.
Maximal tolerierbare Dosis (MTD):
Höchste Dosis im Tierexperiment, bei der keine gravierenden toxischen Effekte allgemeiner Art auftreten. Die MTD wird in der Regel anhand der Körpergewichtsentwicklung ermittelt. In Tierstudien, mit denen die mögliche krebserzeugende Wirkung einer Prüfsubstanz untersucht wird, sollte die MTD erreicht, aber nicht überschritten werden.
Maximum-Likelihood-Schätzung:
Statistisches Verfahren zur möglichst genauen Schätzung der höchsten Wahrscheinlichkeit als Kennwerte für die Grundgesamtheit (Population) auf Basis der vorliegenden Stichprobe.
Mesotheliom:
Bösartiger Tumor des Bauchfells (Peritoneum), des Brust-/ Rippenfells (Pleura) oder des Herzbeutels (Pericard). Pleuramesotheliome des Menschen werden überwiegend durch eingeatmete biobeständige Fasern (Asbest) bestimmter Abmessungen verursacht.
Mitose:
Kernteilung, bei der aus einem Zellkern zwei Tochterkerne mit gleichem Erbgut entstehen.
Mitotischer Prozess:
siehe "Mitose"
MoE:
Abkürzung für "margin of exposure" (siehe dort)
Multi-Hit-Modell:
Dosis-Häufigkeits-Modell, das zur Modellierung von Dosis-Wirkungs-Beziehungen (siehe dort) krebserzeugender Substanzen eingesetzt werden kann und auf der Vorstellung basiert, dass für die Entstehung eines Tumors mehrere Schadensereignisse ("Treffer") notwendig sind.
Multistage-Verfahren, linearisiertes:
Risikoschätzverfahren, das lange Zeit von der US-amerikanischen Umweltbehörde EPA propagiert wurde. Die zu Grunde liegende mathematische Modellfunktion (Multistage-Modell) beschreibt einen Mehrstufenprozess, der zur Ausbildung von klinisch manifesten Tumoren vorausgesetzt wird. Sie dient zur Modellierung der Dosis-Wirkungs-Beziehung (siehe dort) mittels der vorhandenen experimentellen Daten bis in den Niedrigdosisbereich hinein. Anhand einer Geraden, die der Steigung der Modellfunktion im Nullpunkt entspricht, werden dann die Risiken bei niedrigen Dosen abgeschätzt.
Nekrose:
Unkontrollierter Zelluntergang
Nephrotoxizität:
Spezifische Giftwirkung auf die Niere
Odds Ratio:
Die Odds Ratio (OR) ist eine Verhältniszahl aus zwei Chancen ('Odds'). Die Chance ist definiert als der Quotient aus Wahrscheinlichkeit und Gegenwahrscheinlichkeit (einer Erkrankung bei gegebener Exposition bzw. einer Exposition bei gegebener Erkrankung). Bei seltenen Erkrankungen gibt die OR näherungsweise an, um wieviel mal häufiger eine Krankheit eintritt, wenn ein spezifischer Risikofaktor vorhanden ist, als ohne dessen Vorhandensein. Odds Ratios werden als Ergebnis von Fall-Kontroll-Studien erhalten (siehe dort). Eine Odds Ratio unter 1 weist auf ein erniedrigtes Risiko hin, eine über 1 auf ein erhöhtes. Zur Beurteilung der Relevanz der Erhöhung einer Odds Ratio ist die Kenntnis des zugehörigen Konfidenz-Intervalls (siehe dort) erforderlich. Die Odds Ratio wird vor allem in Fall-Kontroll-Studien als Schätzer des Relativen Risikos (siehe dort) interpretiert, da letzteres in Fall-Kontroll-Studien nicht berechnet werden kann. Je seltener die Erkrankung ist, umso besser wird das RR durch das OR approximiert.
Ökologische Studien (bzw. Korrelationsstudien):
Diese Studien vergleichen Exposition und Erkrankung auf Gruppenniveau, d. h. für die Exposition bzw. Erkrankung (oder beide) liegen keine individuellen Informationen vor (z. B. Häufigkeit der Durchführung eines bestimmten Produktionsverfahrens und Krebsmortalität im Vergleich zweier Fabriken). Aufgrund des nicht individuell zugeordneten Expositions- und Erkrankungsstatus sollten ökologische Studien jedoch grundsätzlich nicht für die Ableitung von Expositions-Risiko-Beziehungen zur Einschätzung von Arbeitsplatzgrenzwerten herangezogen werden.
OR:
Odds Ratio (siehe dort)
Parenterale Applikation:
Verabreichung einer Substanz unter Umgehung des Magen-Darm-Trakts (z. B. durch Inhalation oder durch Injektion in eine Vene)
Pfad-zu-Pfad-Extrapolation:
Extrapolation von einem Aufnahme-Pfad zu einem anderen. Am Arbeitsplatz steht die Aufnahme von Arbeitsstoffen über die Atemwege (inhalativ) und die Haut (dermal) im Vordergrund, wahrend in Tierstudien die Prüfsubstanzen häufig verfüttert oder über das Trinkwasser (oral) verabreicht werden. Wegen des teilweise ausgeprägten First-Pass-Effektes (siehe dort) müssen für die Übertragung der Ergebnisse aus Fütterungs-, Trinkwasser- oder Schlundsondenstudien auf Arbeitsplatzverhältnisse mitunter Korrekturfaktoren eingeführt werden.
Peroxisomenproliferation:
Vermehrung von Peroxisomen (Zellorganellen, denen u. a. eine bedeutende Rolle beim Fettstoffwechsel zukommt). Es ist bekannt, dass sich die Peroxisomen der Leber einiger Wirbeltiere, vor allem der Nager, durch die Behandlung mit bestimmten Stoffen ("Peroxisomenproliferatoren", z.B. Fibrate, Phthalate) stark vermehren lassen. Diese Reaktion wird durch einen spezifischen Rezeptor (PPAR(alpha)-Rezeptor) vermittelt, der in der Leber von Nagern ungleich häufiger auftritt als beim Menschen. In Folge der Peroxisomenproliferation können Tumoren in der Nagetierleber induziert werden. Eine Relevanz für den Menschen ist in den meisten Fällen nicht gegeben (siehe auch Nummer 10.3).
Pharmakokinetisches Modell:
Physiologisch basierte pharmakokinetische Modelle (PBPK-Modelle) versuchen, das Verhalten eines Stoffes im Organismus zu beschreiben und die Gewebekonzentrationen in Versuchstier und Mensch zu quantifizieren.
Phäochromocytom:
Tumor des Nebennierenmarks (siehe auch Nummer 10.3)
Point of Departure (POD):
Ausgangswert für weitere Schritte der Risikoabschätzung (siehe "T25- Verfahren")
Population:
In der Epidemiologie wird als Population jede durch mindestens ein Merkmal definierbare Gruppe von Menschen verstanden. Dabei kann es sich um die gesamte Bevölkerung eines Landes oder einer Region handeln oder um eine durch eine spezifische definierte Erkrankung gekennzeichnete Patientengruppe (Patientenpopulation).
Power, statistische:
siehe "Statistische Power"
PPAR(alpha)-Rezeptor:
siehe "Peroxisomenproliferation"
Prämaligne Effekte:
Vorstufen einer bösartigen Neubildung in einem Gewebe
Präputialdrüse:
Drüse im Genitalbereich einiger Säugetiere (z. B. Ratten, Mäuse), die Sexuallockstoffe produziert. Bei weiblichen Tieren spricht man meist von Klitorisdrüse. Der Mensch besitzt kein anatomisches Äquivalent zur Präputial-/Klitorisdrüse.
Prävalenz:
Bezeichnet den Bestand an Patienten mit einer definierten Erkrankung bezogen auf eine definierte Population zu einem bestimmten Zeitpunkt oder kumulativ nach einer bestimmten Beobachtungsdauer einer Population. Sie definiert einen Anteilswert, der üblicherweise als Prozentwert oder als Proportion mit Werten zwischen 0 und 1 angegeben wird.
Primäre Gentoxizität:
siehe "Gentoxisch"
Querschnitts-Studie:
In einer Querschnitts-Studie wird zu einem definierten Zeitpunkt eine definierte Personengruppe, meistens eine Stichprobe der Bevölkerung untersucht. Eine solche Untersuchung erlaubt die Schätzung der Häufigkeit von Merkmalen, Verhaltensweisen, Risikofaktoren (siehe dort). Diese Häufigkeiten werden mit dem epidemiologischen Terminus "Prävalenz" (siebe dort) bezeichnet. Neben der Schätzung von Prävalenzen ist es auch möglich, Mittelwerte von Messwerten (z. B. systolischer Blutdruck, Cholesterinspiegel) zu schätzen. Für beide Ansätze ist in der Planung einer Querschnitts-Studie die Berechnung des erforderlichen Stichprobenumfangs unumgänglich; siehe Stichproben-Berechnung.
Ein Synonym für Querschnitts-Studie ist die Bezeichnung Survey. Querschnitts-Studien stellen eines der bedeutsamsten Instrumente dar, um den Gesundheitszustand einer Bevölkerung zu untersuchen. Nach dem Stand der Wissenschaft müssen Surveys als repräsentative Surveys durchgeführt werden, d. h. auf der Basis einer repräsentativen Zufallsstichprobe aus der Bevölkerung.
REACH:
Bei REACH (Registration, Evaluation, Authorisation and Restriction of Chemicals - Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe) handelt es sich um die grundlegende Verordnung im Rahmen des EU-Chemikalienrechts, mit dem eine europaweite Vereinheitlichung erreicht werden soll. Sie wurde am 18.12.2006 endgültig verabschiedet und trat zum 1.6.2007 in Kraft (EG-Verordnung 1907/2006, Richtlinie 2006/121/EG; EU, 2006).
In sog. "REACH Implementation Projects" (RIP) werden von Arbeitsgruppen auf europäischer Ebene die Methoden und Leitfäden für die Umsetzung der REACH-Verordnung vorbereitet.
Relatives Risiko:
Faktor der angibt, wieviel mal häufiger (bzw. seltener) ein bestimmtes Ereignis (Erkrankung, Tod) in einer Population auftritt im Vergleich zu einer Vergleichspopulation. Das Relative Risiko für Zigarettenraucher, an einem Bronchial-Karzinom zu versterben, ist - je nach der täglich gerauchten Anzahl von Zigaretten und nach der lebenslangen Anzahl von gerauchten Packungen - bis zu 25, d. h. ein starker Raucher hat ein 25-mal größeres Risiko, an Bronchial-Karzinom zu versterben, als ein Nichtraucher. Das Relative Risiko kann bei seltenen Erkrankungen im Rahmen von Fall-Kontroll-Studien mittels des Odds Ratio (siehe dort) zuverlässig geschätzt werden. Diese Bedingung ist bei Krebserkrankungen in aller Regel erfüllt.
Das Relative Risiko (RR) lässt sich definieren als Quotient aus der Inzidenz unter Exponierten (I1) und der Inzidenz unter nicht Exponierten (10):
RR = I1/10
RIP:
REACH Implementation Project, siehe "REACH"
Risiko:
Nach der gesellschaftspolitisch-juristischen Definition (siehe Abschn. IArt. 2 der EU-Richtlinie 98/24/EG) wird unter Risiko im vorliegenden Zusammenhang die Wahrscheinlichkeit des Eintritts einer Krebserkrankung durch die Exposition gegenüber krebserzeugenden Gefahrstoffen verstanden. Bei zunehmender Schadstoffdosis oder Expositionskonzentration eines krebserzeugenden Stoffes erhöht sich das Risiko bzw. die Wahrscheinlichkeit eines Schadenseintritts nimmt zu.
Risikofaktor:
Eigenschaften von Personen bzw. äußere Einwirkungen, die zur positiven oder negativen Beeinflussung eines Krankheitsrisikos/Mortalitätsrisikos führen können. So ist Zigarettenrauchen ein Risikofaktor für die Entstehung von u. a. Bronchial-Karzinomen, Bronchitis, Myokardinfarkt, Magen- und Blasen-Karzinomen, Leukämien. Die LDL-Fraktion des Cholesterins ist ein Risikofaktor für die Entstehung arteriosklerotischer Veränderungen, während die HDL-Fraktion des Cholesterins als "positiver" Risikofaktor offensichtlich die Entstehung von Myokardinfarkten verhindern kann. Manche Wissenschaftler betrachten auch das Geschlecht und das Alter einer Person als Risikofaktoren. Berufliche Einwirkungen, Umweltfaktoren und sozio-ökonomische Charakteristika haben sich für eine Vielzahl von Erkrankungen als starke Risikofaktoren erwiesen.
Risikozahl:
Die Risikozahl stellt im vorliegenden Zusammenhang einen unter bestimmten Annahmen berechneten Wert für das expositionsbedingte Lebenszeitrisiko im Szenario einer Exposition über das gesamte Arbeitsleben dar. Das Lebenszeitrisiko gibt die Wahrscheinlichkeit an, im Laufe des Lebens an einer bestimmten Krebsart zu erkranken, wenn die Sterblichkeit an anderen Ursachen ungefähr gleich ist wie in einer nichtexponierten Population. Die Risikozahl kann auch als Schätzung des Exzess-Risikos (siehe dort) bzw. als "additional risk" (siehe, dort) oder "extra risk" (siehe dort) bezeichnet werden, da dabei das Hintergrundrisiko entsprechend in Anrechnung gebracht wurde.
RR:
Relatives Risiko (siehe dort)
Schätzen:
Unbekannte Parameter der Grundgesamtheit werden anhand von Beobachtungswerten aus einer Stichprobe angenähert. Dafür stehen verschiedene statistische Verfahren zur Verfügung. So werden Populationsmittel durch Stichprobenmittelwerte geschätzt. Um diese Punktschätzer besser beurteilen zu können, wird deren Unscharfe anhand der in der Stichprobe geschätzten Variabilität des jeweiligen Merkmals in der Population beurteilt. Zur besseren Beurteilung von Punktschätzern wie z. B. geschätzte Relative Risiken (RR), werden diese und ihre Variabilitätsschätzer in so genannten Konfidenzintervallen zusammengeführt, die mit einer vorgegebenen Sicherheitswahrscheinlichkeit, z. B. 95 Prozent, grob gesprochen Aussagen wie "das RR liegt mit 95-prozentiger Wahrscheinlichkeit zwischen 2,0 und 5,5" erlauben.
siehe "Wirkungsschwelle, toxikologische"
Sekundäre Gentoxizität:
siehe "Gentoxisch"
Sicherheitsfaktor:
siehe "Extrapolationsfaktor/Sicherheitsfaktor"
SIR:
Standardisierte Inzidenzratio (siehe dort)
SMR:
Standardisierte Mortalitätsratio (siehe dort)
Standardisierte Inzidenzratio (SIR):
Anzahl der in einem bestimmten Zeitraum beobachteten Neuerkrankungsfälle in einer Studienpopulation dividiert durch die Anzahl von Neuerkrankungsfällen, die erwartet würde, wenn die altersspezifischen Inzidenzraten (siehe "Inzidenz") der Studienpopulation dieselben wären wie die altersspezifischen Inzidenzraten einer externen Vergleichspopulation.
Standardisierte Mortalitätsratio (SMR):
Anzahl der in einem bestimmten Zeitraum beobachteten Todesfälle (einer bestimmten Ursache) in der Studienpopulation dividiert durch die Anzahl von Todesfällen, die erwartet würde, wenn die altersspezifischen Mortalitätsraten der Studienpopulation dieselben wären wie die altersspezifischen Mortalitätsraten einer externen Vergleichspopulation.
Statistische Power:
Wahrscheinlichkeit, mit der ein statistischer Test (tatsächlich vorhandene) Unterschiede (z. B. unterschiedliche Tumorraten bei exponierten gegenüber nicht exponierten Versuchstieren) aufdecken und von zufälligen Schwankungen abgrenzen kann. Die statistische Power ist u. a. abhängig vom Umfang der Stichprobe (Anzahl von Versuchstieren in einer Dosisgruppe). Mit dieser Größe kann somit abgeschätzt werden, wie groß eine Studienpopulation sein muss, um ermittelte Unterschiede statistisch abzusichern und Zufallseffekte auszuschließen (siehe auch "Stichprobenumfangs-Berechnung").
Sterbetafelmethode:
Statistisches Verfahren zur Berechnung des Lebenszeitrisikos, an einer bestimmten Krebsart zu sterben. Bei der Sterbetafelmethode werden die altersspezifischen Mortalitätsraten für die Krebsart und für alle Todesursachen zur Berechnung des Lebenszeitrisikos benutzt.
Stichprobenumfangs-Berechnung:
Die Planung jeder epidemiologischen Studie erfordert die Berechnung der Stichprobengröße, die nötig ist, um diejenigen Annahmen zu verifizieren oder zu falsifizieren, die der Untersuchungshypothese zu Grunde liegen. Zur Berechnung des Stichprobenumfangs sind verschiedene Festlegungen erforderlich:
- 1.
Das Signifikanz-Niveau oder die Wahrscheinlichkeit für den Fehler I. Art: Hier wird festgelegt, mit welcher statistischen Sicherheit ein möglicher Unterschied (beim Vergleich mehrerer Gruppen) oder eine Risiko-Erhöhung berechnet werden soll. Üblich ist es, das Signifikanz-Niveau auf höchstens fünf Prozent festzulegen. Je kleiner das Signifikanz-Niveau ist, umso größer muss die Stichprobe ausfallen. Wird in einer Berechnung ein Signifikanzniveau von fünf Prozent erreicht oder unterschritten, so bedeutet dieses, dass in mindestens 95 Prozent aller denkbaren vergleichbaren Studien tatsächlich der hier berechnete Unterschied auch auftreten wird.
- 2.
Die Power oder die Wahrscheinlichkeit des Fehlers II. Art: Hierbei wird festgelegt, in wie viel Prozent aller denkbaren Konstellationen ein tatsächlich vorhandener Unterschied oder eine bestehende Risiko-Erhöhung nicht übersehen wird. Eine Power von 90 Prozent würde also bedeuten, dass das Risiko, einen Unterschied nicht zu entdecken - obgleich ein Unterschied vorhanden ist - nicht größer als zehn Prozent ist. Wünschenswert ist es natürlich, die Power einer Studie möglichst groß zu machen. Je größer die Power ist, umso größer muss auch die Stichprobe ausfallen. Bei epidemiologischen Studien sollte die Power nicht kleiner als 80 Prozent angesetzt werden.
- 3.
Annahmen über die minimale Größe einer Risiko-Erhöhung: Je kleiner das zu entdeckende Risiko ist, umso größer muss die Stichprobe sein, um die Risikoerhöhung bei einer vorgegebenen Power noch zu entdecken. Für die Festlegung dieses Parameters kann man entweder auf vorgehende Untersuchungen zurückgreifen oder es müssen plausible Annahmen zu Grunde gelegt werden. Im Umweltbereich sind Risiko-Erhöhungen über 100 Prozent relativ selten anzutreffen.
- 4.
Annahmen über die Häufigkeit eines kritischen Risikofaktors in der Vergleichsgruppe bzw. Vergleichspopulation: Sollen in einer Studie mehrere Risikofaktoren gleichzeitig analysiert werden, empfiehlt es sich, für die Stichproben-Berechnung den seltensten Risikofaktor zu Grunde zu legen. Stehen keine exakten Daten über die Häufigkeit von Risikofaktoren zur Verfügung, empfiehlt sich die Durchführung einer Pilotstudie. Hilfsweise können auch Informationen aus publizierten Studien herangezogen werden.
Stratum (Mehrzahl: Strata):
In der Epidemiologie: Untergruppe eines Kollektivs. Die Unterteilung einer Studienpopulation in Untergruppen (z. B. nach Alter, Geschlecht, Rauchgewohnheiten) bezeichnet man als Stratifizierung.
Stratifizierung:
siehe "Stratum"
Sublinearität:
siehe "Dosis-Wirkungs-Beziehungen"
Supralinearität:
siehe "Dosis-Wirkungs-Beziehungen"
T25:
Tumorgene Dosis, die 25 Prozent zusätzliche Inzidenz erwarten lässt. Ursprünglich wird die T25 im experimentellen System als Dosis (mg/kg x d) angegeben. Im vorliegenden Rahmen werden auch Transformationen in eine Inhalationskonzentration als T25 oder hT25 (siehe dort) bezeichnet (siehe auch T25-Verfahren/-Methode).
T25-Verfahren:
Einfaches Risikoabschätzungsverfahren, das von der Europäischen Kommission zur Ableitung von Grenzwerten für Zubereitungen mit krebserzeugenden Stoffen empfohlen wurde (EC, 2002; Dybing et al, 1997; Sanner et al, 1997). Ausgehend von einer Konzentration mit signifikant erhöhter Tumorinzidenz wird durch lineare Interpolation
- 1.
unter Berücksichtigung der Hintergrundinzidenz,
- 2.
gegebenenfalls unter Korrektur einer nicht lebenslangen Versuchsdauer, und
- 3.
unter Annahme einer vollständigen Resorption eine Dosis ermittelt, bei der die Inzidenz für diesen Tumor im Tierversuch 25 Prozent bei lebenslanger Exposition beträgt.

Der T25-Wert kann dann als "point of departure" (Ausgangspunkt) verwendet werden, um durch lineare Extrapolation in den Niedrigdosisbereich das Risiko für geringere Dosierungen abzuschätzen (vgl. Abbildung).
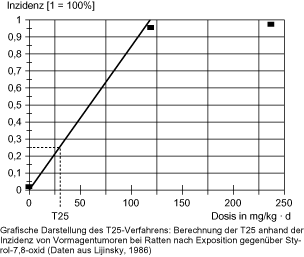
Bei dem T25-Verfahren wird die tatsächliche Dosis-Wirkungsbeziehung und die Streubreite der experimentellen Daten nicht berücksichtigt, da zur Berechnung der tumorigenen Dosis 25 Prozent nur die Hintergrundinzidenz und die Inzidenz bei einer Expositionskonzentration herangezogen werden.
Tolerables Risiko:
siehe "Akzeptables/tolerables Risiko"
Topoisomerasen:
Enzyme, die den spiralisierten DNA-Doppelstrang entwinden können und eine wichtige Rolle bei Zellteilung und Eiweißsynthese spielen.
Toxikodynamik:
Lehre von der Wirkung giftiger Stoffe auf den Organismus (siehe auch Toxikokinetik)
Toxikokinetik:
Lehre vom Schicksal giftiger Substanzen im Organismus (Aufnahme, Verteilung, Stoffwechsel, Ausscheidung) (siehe auch Toxikodynamik)
Vormagen:
Drüsenloses Verdauungsorgan vor dem Hauptmagen von Nagetieren. Nach Verabreichung von gentoxischen Kanzerogenen (siehe "Gentoxizität") mit dem Futter oder per Schlundsonde an Nagetiere entstehen häufig Vormagentumoren. Der Mensch besitzt keinen Vormagen.
Wirkungsschwelle, toxikologische:
Im Allgemeinen versteht man unter einer toxikologischen Wirkungsschwelle eine Dosis oder Expositionskonzentration (Schwellenwert), bei deren Unterschreitung ein bestimmter Effekt nicht auftritt. Der Begriff ist nicht zu verwechseln mit dem "no observed effect level" (NOEL), der eine signifikante beobachtete Effekterhöhung gegenüber einem "Hintergrund" angibt und vom jeweiligen Studiendesign abhängt.
Ebenso wie es eine Vielzahl von Definitionen zur toxikologischen Wirkschwelle gibt, ist es umstritten, ob bei einzelnen Schritten der Krebsauslösung durch chemische Kanzerogene Wirkschwellen existieren (Neumann 2006a, b, c). Für "epigenetische" nicht-gentoxische Kanzerogene (z.B. zytotoxische [siehe Zytotoxizität], immunschädigende Substanzen oder hormonähnliche Wachstumsstimulatoren) wird in der Regel eine Wirkschwelle angenommen. Es wird aber auch diskutiert, ob auf bestimmte sekundär gentoxische (siehe dort) Kanzerogene das Modell einer Wirkschwelle angewendet werden kann (Hengstler et al, 2006). Auch wenn es Argumente für eine solche Ansicht gibt, erscheint die experimentelle Detektion eines Schwellenwerts in diesen Fällen schwierig.
Zellproliferation:
Vermehrung von Zellen in einem Gewebe
Zymbaldrüse:
Talgdrüse im äußeren Gehörgang von Nagetieren. Der Mensch besitzt keine Zymbaldrüsen.
Zytotoxizität:
Schädigende Substanzwirkung auf Gewebezellen
Literatur:
- [1]
Dybing, E.; Sanner, T.; Roelfzema, H.; Kroese, D.; Tennant, R.W., 1997 T25: a simplified carcinogenic potency index: description of the System and study of correlations between carcinogenic potency and species/site specificity and mutagenicity Pharmacology & Toxicology, 80,1997, 272-279
- [2]
EC, European Commission, 2002 Guidelines for Setting Specific Concentration Limits for Carcinogens in Annex I of Directive 67/548/EEC. Inclusions of Potency Considerations Commission Working Group on the Classification and Labelling of Dangerous Substances, http://ecb.jrc.it/classification-labelling/, 2002
- [3]
Hengstler, J.G.; Degen, G.H.; Foth, H.; Bolt, H.M., 2006 Thresholds for specific classes of genotoxic carcinogens: a new strategy for carcinogenicity categorisation of chemicals to be published in SIIC, in press, 2006
- [4]
Lijinsky, W., 1986 Rat and mouse forestomach tumors induced by chronic oral administration of styrene oxide Journal of the National Cancer Institute, 77, 1986, 471 - 476
- [5]
Neumann, H.G., 2006a Die Risikobewertung von Kanzerogenen und die Wirkungsschwelle, Teil II Bundesgesundheitsblatt, 49, 2006, 665-674
- [6]
Neumann, H.G., 2006b Die Risikobewertung von Kanzerogenen und die Wirkungsschwelle, Teil II Bundesgesundheitsblatt, 49, 2006, 818-823
- [7]
Neumann, H.G., 2006c Die Risikobewertung von Kanzerogenen und die Wirkungsschwelle, Teil III Bundesgesundheitsblatt, 49, 2006, 911-920
- [8]
Sanner, T.; Dybing, E.; Kroese, D.; Roelfzema, H.; Hardeng, S., 1997 Potency grading in carcinogen Classification Molecular Carcinogenesis, 20,1997,280-287
10.2 Berechnungsbeispiele
Beispiel 1: Trichlorethylen - zu Nummer 5.2
Vorbemerkung: Zu Trichlorethylen wurde durch den Unterausschuss III des Ausschusses für Gefahrstoffe eine Bewertung vorgenommen, die eine mechanistische Diskussion und eine quantitative Diskussion der Wirkung beinhaltete. Deshalb wurde dieses Beispiel unter Berufung auf die regulatorische Bewertung in einem deutschen Gremium ausgewählt. Es ist den Autoren des Leitfadens bewusst, dass für die meisten Stoffe dennoch widersprüchliche Einschätzungen zur Datenlage und deren Bewertung (Wirkungsmechanismus, Validität epidemiologischer oder tierexperimenteller Daten, quantitative Schlussfolgerungen) vorliegend Dies trifft auch für Trichlorethylen zu. Das Stützen auf die Beschlusslage des Ausschusses für Gefahrstoffe und seiner Gremien setzt somit Voraussetzungen und ermöglicht die Beschäftigung mit der Berechnungsmethode für nichtlineare Expositions-Risiko-Beziehungen, ohne dass die Berechtigung der gewählten Voraussetzung (also die Beschlusslage zu TRI und deren Begründung) an diesem Ort thematisiert werden sollen.
Trichlorethylen (TRI) wird in Deutschland insbesondere wegen der bei hoher beruflicher Exposition beobachteten Nierenkrebsfälle als Humankanzerogen eingeordnet, jedoch wird aus hier nicht näher zu erläuternden Gründen angenommen, dass eine zytotoxische Wirkung auf die Niere maßgeblich zum Krebsgeschehen beiträgt. Eine lokale gentoxische Wirkung in der Niere ist nicht auszuschließen, so dass keine eindeutige Wirkschwelle für TRI ermittelt werden kann. Damit bietet sich TRI als Beispiel für Absatz 4 in Nummer 5.1 an. Die folgende Ausführung des Beispiels soll bei realen Daten jedoch nur den Rechenprozess erläutern, ohne den Anspruch auf eine weitergehende Dokumentation und Diskussion der stoffspezifischen Informationen zu erheben. Zusätzlich neben den hier referierten Daten liegen umfangreiche weitere Studien zum Wirkmechanismus, zur Gentoxizität, zur Nephrotoxizität, zur Kanzerogenität in der Niere und zur Kanzerogenität und Toxizität in anderen Organen vor, die jedoch nicht ausgeführt werden.
Roller (2005) leitete auf Basis der deutschen Studien zu Nierenkrebs nach beruflicher Exposition gegenüber Trichlorethylen ein Exzess-Risiko von ca. 5 Prozent bei einer Exposition gegenüber 100 ppm (mit Spitzen gegenüber 500 ppm) ab (18 Jahre Exposition, 2 h/d, 3 d/Wo. Spitzenexposition, sonst ca. 100 ppm). Insgesamt werden in der Berechnung 3.000 ppm-Jahre Exposition zu Grunde gelegt.
Expositionshöhe
Für die Studien, bei denen in Deutschland erhöhte Nierenkrebsrisiken festgestellt wurden, wird zumindest bei einem beträchtlichen Teil der Arbeitsplätze von sehr hohen Expositionen ausgegangen, die auch zu pränarkotischen Symptomen geführt haben. Daraus lässt sich schließen, dass Konzentrationen von 200 ppm häufig überschritten wurden. Es wird angenommen, dass die Konzentrationen für 2-3 Stunden an 2-3 Tagen pro Woche ungefähr 500 ppm betragen haben mögen. Für die Studie von Henschler et al (1995) ist eine durchschnittliche Beschäftigungsdauer von ungefähr 18 Jahren anzusetzen. Geht man ferner davon aus, dass auch in der Zeit, in der keine Spitzenexpositionen erreicht wurden, noch TRI-Expositionen vorhanden waren, dann lässt sich folgende Exposition abschätzen:
500 ppm, 2 h/d, 3 d/Wo., 18 Jahre
plus 100 ppm, 6 h/d, 3 d/Wo., 18 Jahre
plus 100 ppm, 8 h/d, 2 d/Wo., 18 Jahre.
Insgesamt entspricht dieses Szenario einer regelmäßigen vollschichtigen Exposition für 18 Jahre gegenüber mindestens 100 ppm, wobei wiederholt in jeder Woche Expositionsspitzen von 500 ppm für längere Zeit auftraten. Gerundet ergibt sich rechnerisch für dieses Expositionsszenario ein Wert von 3.000 ppm-Jahren für die kumulative Exposition.
Risikozuordnung (Exzess-Risiko)
Die Erhöhungen des Nierenkrebsrisikos, die in den deutschen Studien zur TRI-Exposition beobachtet wurden, variieren etwas in Abhängigkeit vom Untersuchungszeitraum und von der Definition des Merkmals "exponiert".
Die Odds-Ratio-Werte (OR) in den Fall-Kontroll-Studien liegen meist statistisch signifikant im Bereich um 2 oder 3, wobei auch höhere Werte auftreten (z. B. "any exposure in metal degreasing" OR = 5,57 bei Brüning et al, 2003), das höchste OR wurde in der Studie von Vamvakas et al. (1998) mit 10,8 festgestellt. Für die Risikobewertung ist die Umrechnung der in den Studien genannten Maßzahlen des "Relativen Risikos" (insbesondere OR) in Zahlenwerte des "Absoluten Risikos" erforderlich. Informationen zur Krebsmortalität in der Allgemeinbevölkerung lassen sich der Datenbank der WHO (http://www.who.int/whosis/en/ ) entnehmen. Danach betrug in Deutschland der Anteil der Todesursache "Bösartige Neubildung der Niere, ausgenommen Nierenbecken" (ICD/9 189.0) an allen Todesursachen im Jahre 1990 bei den Männern 0,66 Prozent (2811/425093), bei den Frauen 0,42 Prozent (2085/496352); im Jahre 1997 betrugen die Anteile 0,77 bzw. 0,48 Prozent (WHO, 2003). Nach diesen Zahlen muss von einem Lebenszeit-Mortalitätsrisiko für Nierenkrebs in der männlichen Allgemeinbevölkerung in Deutschland in Höhe von zirka 0,7 Prozent ausgegangen werden.
Eine Verdoppelung dieses Risikos (RR, SMR oder OR von 2,0) bedeutet ein zusätzliches (Exzess-)Lebenszeitkrebsrisiko in derselben Höhe.
Bei den angegebenen Zahlen handelt es sich um Mortalität, das eigentlich zu betrachtende Inzidenzrisiko ist höher. Präzise Daten über die Nierenkrebsinzidenz in der gesamten Bundesrepublik Deutschland liegen nicht vor, die Publikation "Krebs in Deutschland" (2004) enthält jedoch datengestützte Schätzungen der Inzidenzraten. Für das Jahr 2000 sind dort die geschätzten Inzidenzraten und die Mortalitätsraten gemäß amtlicher Statistik für Nierenkrebs einander gegenübergestellt. Die Raten bei den Männern betragen demnach 22,0 (Inzidenz) bzw. 9,7 (Mortalität) pro 100.000 und Jahr, bei den Frauen liegen die entsprechenden Zahlenwerte bei 15,0 bzw. 6,2.
Als Verhältnis von Inzidenz zu Mortalität ergibt sich demnach ein Wert von rund 2,3.
Wendet man diesen Faktor auf das Mortalitätsrisiko von 0,7 Prozent an, dann erhält man den Wert von 1,6 Prozent für das absolute Basis-Inzidenzrisiko für Nierenkrebs der Männer in den 90er-Jahren in Deutschland. Selbstverständlich sind die Odds-Ratio-Werte der epidemiologischen Studien zu Nierenkrebs nach TRI-Exposition mit Unsicherheiten behaftet, insgesamt ist es jedoch unzweifelhaft, dass eine signifikante Erhöhung des Nierenkrebsrisikos aufgrund eines ursächlichen Zusammenhangs mit der Exposition dort nur wahrscheinlich ist, wenn dieser Signifikanz ein ExzessInzidenzrisiko im Prozentbereich entspricht. Bereits ein Relatives Risiko von 2,0 bedeutet bei einem Basisrisiko von 1,6 Prozent ein Exzess-Risiko von ebenfalls 1,6 Prozent.
Es erscheint daher gerechtfertigt, der sehr hohen kumulativen Exposition von 3.000 ppm-Jahren ein Exzess-Nierenkrebsrisiko von fünf Prozent zuzuordnen.
Risikoextrapolation (linear)
Ausgehend von dieser Berechnung verwenden wir im Folgenden ein Exzess-Risiko von fünf Prozent bei Exposition über 3.000 ppm-Jahre als "point of departure". Da Humandaten mit Risikoangaben bei geringerer Inzidenz als 25 Prozent vorliegen, ist nach Nummer 3.7 Abs. 2 keine Umrechnung auf einen T25 oder HT25 sinnvoll. 3.000 ppm-Jahre über ein ganzes Arbeitsleben von 40 Jahren lassen sich auch auf eine durchschnittliche Exposition von 75 ppm (x 40 Jahre) umrechnen. Würden wir ausgehend von dieser Angabe eine lineare Extrapolation durchführen, ergäbe sich somit ein Risiko von
Durchschnittlich ppm ppm-Jahre (40Jahre Exposition) Exzess-Risiko Bemerkung 75 ppm 3.000 5 % POD; deutsche epidemiologische Studien zu Nierenkrebs 15 ppm 600 1 % linear 6 ppm 240 0,4 % Linear; bei Wirkschwelle für nichtkanzerogene Nephrotoxizität bei Exposition gegenüber TRI 1,5 ppm 60 0,1 % Linear 60pph 2,4 0,004 % Linear Das Exzess-Risiko ist somit bei angenommener Linearität durch folgende Gleichung zu beschreiben:
Exzess-Risiko [Prozent] = 0,067 x Konzentration [ppm] für alle Bereiche bei und unter 75 ppm
Risikoextrapolation (nichtlinear)
Nach Beobachtungen von Green et al. (2004) wurden bei im Mittel 32 ppm Expositionshöhe bei TRI-exponierten Arbeitern noch signifikant subklinische Niereneffekte gefunden. Die Biomarker für subklinische Nephrotoxizität waren bei 23 Arbeitern, die gegenüber 6 ppm TRI mehrjährig exponiert waren, nicht mehr erhöht (Seiden et al, 1993). Angesichts der nur geringen Effektstärke bei 32 ppm kann der NOAEL bei 6 ppm ohne weitere Extrapolationsschritte als Schwelle für Nephrotoxizität auch bei großen Kollektiven herangezogen werden. Wir verwenden daher die Konzentration von 6 ppm als TC* und nehmen an, dass an diesem Punkt das Risiko um eine Größenordnung niedriger ist, als durch die lineare Berechnung (siehe oben, Tabelle) ermittelt. Dadurch ergibt sich für 6 ppm ein Risiko (neu) von 0,04 Prozent und eine Expositionsrisikogleichung von
Exzess-Risiko [Prozent] = 0,072 x Konzentration [ppm] - 0,39
für den Bereich zwischen Konzentration [6ppm, 75 ppm]
Exzess-Risiko [Prozent] = 0,0067 x Konzentration [ppm] für den Bereich mit Konzentrationen [< 6ppm]
Durchschnittlich ppm ppm-Jahre Exzess-Risiko Bemerkung 75 ppm 3.000 5 % POD; deutsche epidemiologische Studien zu Nierenkrebs 19,3 ppm 772 1 % linearisiert ("steiler" Teil) 6,8 ppm 272 0,1 % linearisiert ("steiler" Teil) 6 ppm 240 0,04 % "Knickpunkt"; bei Wirkschwelle für nichtkanzerogene Nephrotoxizität bei Exposition gegenüber TRI 1,5 ppm 60 0,01 % linearisiert ("flacher" Teil) 0,6 ppm 24 0,004 % linearisiert ("flacher" Teil) Das nominelle Risiko von 1:1.000 läge z. B. nach der linearen Extrapolation bei 1,5 ppm, während es bei ca. 7 ppm liegt, wenn eine Nichtlinearität begründet angenommen werden kann. Unter 6 ppm zeigt sich ein im Wesentlichen um eine Größenordnung reduziertes Risiko gegenüber dem Linearansatz.
Die folgende Abbildung stellt das Ergebnis im unteren ppm-Bereich graphisch dar:
Literatur
- [1]
Brüning, T.; Pesch, B.; Wiesenhütter, B.; Rabstein, S.; Lammert, M.; Baumüller, A.; Bolt, H.M., 2003 Renal cell Cancer risk and occupational exposure to trichloroe-thylene: results of a consecutive case-control study in Arnsberg, Germany American Journal of Industrial Mediane, 43, 2003, 274-285
- [2]
Green, T.; Dow, J.; Ong, C.N.; Ng, V.; Ong, H.Y.; Zhuang, Z.X.; Yang, X.F.; Bloemen, L., 2004 Biological monitoring of kidney function among workers occupationally exposed to trichloroethylene Occupational and Environmental Medicine, 61, 2004, 312-317
- [3]
Henschler D, Vamvakas S, Lammert M, Dekant W, Kraus B, Thomas B, Ulm K., 1995 Increased incidence of renal cell tumors in a cohort of cardbord workers exposed to trichloroethylene. Arch Toxicol 69: 291-299,1995
- [4]
Seidén, A.; Hultberg, B.; Ulander, A.; Ahlborg, G., 1993 Trichloroethylene exposure in vapour degreasing and the urinary excretion of N-acetyl-ß-d-glucosaminidase Archives of Toxicology, 67,1993, 224-226
- [5]
Vamvakas, S.; Brüning, T.; Thomasson, B.; Lammert, M.; Baumüller, A.; Bolt, H.M.; Dekant,. W.; Birner, G.; Henschler, D.; Ulm, K., 1998 Renal cell Cancer correlated with occupational exposure to trichloroethylene Journal of Cancer Research and Clinical Oncology, 124, 1998, 374-382
- [6]
WHO (World Health Organization), 2003 WHO Statistical Information System (WHOSIS). WHO Mortality Data Base. Global Programme on Evidence for Health Policy Assessing Health Needs: Epidemiology and Bürden of Disease Unit. Genf: World Health Organization. Last updated September 2003. http://www3.who.int/whosis/menu.cfm
Beispiel 2:
Zu Nummer 5.3 (Schwellenwert), theoretisches Beispiel
Beispiel:
Bei einem Stoff A tritt bei einer Raumluftkonzentration von 200 mg/m Krebs im Respirationstrakt bei 3/50 Tieren (Ratte) auf bei 50 mg/m3 bei 0/50 Tieren, bei der Kontrollgruppe ebenfalls bei 0/50 Tieren. (Expositionsmuster 6h/d; 5d/w; 104 Wochen, Nachbeobachtung auf Lebenszeit). Als Mechanismus für die krebserzeugende Wirkung sei eine reine Sekundärreaktion auf eine Irritation der Atemwege mit einem NOAEL (90 Tage) von 100 mg/m3 ausreichend belegt. Als Extrapolationsfaktoren sind nach dem DNEL-Konzept heranzuziehen (Annahme, dass keine weiteren Korrekturen gegenüber dem Default angemessen begründbar sind): Zeitextrapolation: 2; Interspeziesextrapolation (Variabilität): 2,5; Intraspeziesextrapolation: 5; Zusätzlicher Faktor wegen der Schwere der beobachteten sekundären Tumorgenität: 10. Der Gesamtextrapolationsfaktor beträgt damit 25 bzw. 250. Der NOAEL entspricht einer humanäquivalenten Lebenszeitexposition von 50 mg/m3 bei leichter Aktivität und täglich 8h Exposition ( Nummer 4.2). Nach Korrektur auf Lebensarbeitszeit (x 75/40) ergibt sich der NOAEL zu 93,75 mg/m3. Damit ergibt sich ein T* von 93,75 /25 = 3,75 mg/m3 ~ 4 mg/m3 bzw. ein T*/10 von 0.4 mg/m. Regulatorisch würden somit als Wirkschwelle 0,4 mg/m3 für den Arbeitsplatz herangezogen. Wäre nur Reizung (kein Krebs) beobachtet worden, so würden 2 mg/m im Default als DNEL berechnet (keine Korrektur für Exposition über Lebenszeit/Lebensarbeitszeit bei DNEL-Konzept für Nichtkanzerogene). Wir gehen in diesem Beispiel von einem T25 von 833 mg/m3 aus. Gegenüber dem T25 liegt diese angenommene Wirkschwelle bei linearer Extrapolation bei ca. bei 0,01 Prozent (1:10.000). (Das (theoretische) Beispiel demonstriert auch, dass es Datensätze geben kann, bei denen die Differenzierung zwischen linearer Extrapolation, nichtlinearer Extrapolation und Schwellenwert nicht zu gravierenden quantitativen Unterschieden führt).
Beispiel Butadien
(Grundlage: AGW-Begründung/Positionspapier des Arbeitskreises "Grenzwerte und Einstufungen für CM-Stoffe" (AK CM) im UA III des AGS zu 1,3-Butadien)
1. Systematische Literatursuche
Der Bewertung voraus geht eine strukturierte systematische Literatursuche. Folgende Studien zur industriellen BD-Exposition und dem Risiko einer Krebserkrankung wurden identifiziert:
Für eine nordamerikanische Kohorte von Arbeitern in der Kunstgummi-Produktion liegen zahlreiche publizierte Ergebnisse mit detaillierten Expositionsschätzungen unter Angabe der absoluten Butadienkonzentration vor. Diese beziehen sich auf unterschiedliche Follow-up-Zeitpunkte der Kohorte bzw. wurden mit unterschiedlichen Quantifizierungskonzepten der Exposition bzw. unterschiedlichen statistischen Methoden berechnet. Für den Umgang mit Butadien ist die Mortalität an bestimmten Tumoren des lymphatischen bzw. hämatopoetischen Systems erhöht.
Daneben wurden Studien in der Produktion des Butadien-Monomers durchgeführt, für die jedoch keine Absolutangaben zur Exposition (d. h. ppm oder mg/m3) veröffentlicht wurden. Diese Studien können deshalb nicht für die Aufstellung von Expositions-Risiko-Beziehungen herangezogen werden.
Zwei Publikationen mit einem aktuellen Follow-up der Kohorte in der Kunstgummi-Produktion, die zudem eine aktualisierte und verbesserte Job-Expositions-Matrix QEM) für die Expositionsquantifizierung zu Grunde gelegt haben, können als die relevantesten Auswertungen dieser Kohorte angesehen werden. Sie werden deshalb bevorzugt bei der Expositionsabschätzung berücksichtigt (Graff et al., 2005; Cheng et al., 2007). Die eine Publikation berechnet das Risiko mittels einer Poisson-Regression, die zweite Hazard-Rate Ratios mittels Cox-Proportional-Hazards-Regression. Bei Graff sind die Expositionskategorien in Quartile der Exposition unter den Exponierten eingeteilte bei Cheng in Dezile.
Zur Ermittlung von Grenzwerten sollten alle Artikel, die unterschiedliche statistischen Methoden bzw. verschiedene Expositionsmodelle beschreiben, getrennt ausgewertet und kritisch diskutiert werden. Auf eine Meta-Analyse wird verzichtet.
2. Berücksichtigung der Zielparameter
In den ausgewählten Kohortenstudien war die Mortalität an bestimmten Tumoren des lymphatischen bzw. hämatopoetischen Systems erhöht. Die deutlichsten Erhöhungen lassen sich auswerten, wenn die Todesfälle mit den verschiedenen Leukämieformen zu "alle Leukämien" bzw. zu "Leukämie" zusammengefasst werden. Daten zu vorgezogenen Endpunkten anhand biologischer Marker wurden in den Studien nicht veröffentlicht.
Der Einfachheit halber beschränkt sich die folgende Darstellung zur Berechnung der Risikozahl auf die Studie von Graff et al. (2005).
3. Berechnung der Risikozahl
Die folgende Darstellung beschränkt sich auf zwei individuelle Expositionsszenarien: kumulierte ppm-Jahre und ppm-Jahre aufgrund von Expositionsintensitäten von max. 100 ppm.
Tab. 1 zeigt die Expositionsbereiche und dazu errechneten Risikoschätzer aus Graff et al. Bei Graff sind die Expositionskategorien in Quartile der Exposition unter den Exponierten eingeteilt. Da kein Mediane bzw. geometrisches Mittel für die einzelnen Expositionskategorien angegeben ist, wird die Klassenmitte der untersuchten Expositionskategorien zu Grunde gelegt.
Klassenmitte geteilt durch die Dauer der Exposition von 35 Arbeitsjahren(11)liefert den Langzeitmittelwert der Exposition in ppm. Die Klassenmitte für die höchste Expositionskategorie wurde geschätzt.
Die Langzeitmittelwerte werden gegen das Relative Risiko in einem Scatterplot aufgetragen und es wird eine lineare Regressionsgerade berechnet, deren Steigung den Risikoanstieg pro Expositionsunit (ppm BD) ausdrückt (siehe Abb. 1 für die Graff-Studie). Je nach Expositionsmodell ergeben sich Steigungskoeffizienten für das Relative Risiko von 0,16 bzw. 0,31 pro ppm nach 35-jähriger Arbeitsplatzexposition. Die Steigungskoeffizienten der Geraden in Abb. 1a sprechen für die Zuordnung eines Verdoppelungsrisikos (RR=2) bei einem Langzeit-Mittelwert von 5 ppm über einen Zeitraum von 35 bis 40 Jahren (was einer kumulativen Exposition von rund 200 ppm-Jahren entspricht). Der Steigungskoeffizient von Expositionen kleiner gleich 100 ppm ist mit 0,31 pro ppm größer als bei Berücksichtigung aller Expositionswerte (siehe Abb. 1b).
Um diese Information in eine Aussage zum absoluten Lebenszeitrisiko umzuwandeln, ist Information zum Basisrisiko (Hintergrundrisiko) erforderlich. Aufgrund der Mortalität an Leukämie und an allen Ursachen bei der männlichen Allgemeinbevölkerung in den USA und in anderen Industriestaaten lässt sich von einem Lebenszeit-Hintergrundrisiko für Leukämie in Höhe von einem Prozent ausgehen (Roller et al, 2006). Dies bedeutet, dass die Steigungskoeffizienten des Relativen Risikos von 0,16 und 0,31 pro ppm einem Anstieg des absoluten Risikos von 0,16 bzw. 0,31 Prozent pro ppm BD entsprechen. Der untere der beiden Werte bedeutet gerundet ein Exzess-Lebenszeitrisiko in Höhe von 0,2 Prozent (2 zu 1.000) nach einer 35jährigen Arbeitsplatzexposition gegenüber einem Langzeitmittelwert von 1 ppm. Entsprechende Zuordnungen von Exposition und Risikozahl gemäß dem linearen Modell sind in Tabelle 2 für verschiedene Expositionsszenarien enthalten.
4. Abweichende Expositionsmodelle und potenzielle Bias
Zur Risikoabschätzung wurden in den Originalpublikationen verschiedene Modelle berechnet: das hier dargestellte "Single Agent"-Modell, welches nur die Exposition gegenüber BD (adjustiert für Alter und Zeit seit Beschäftigungsbeginn) berücksichtigt bzw. ein multiple Agent Modell, welches mögliches Confounding durch andere Arbeitsstoffe und allgemeine Confounder, wie Styrol und DMDTC berücksichtigt. Für Styrol lässt sich jedoch festhalten, dass die Exposition in der BD-Produktion deutlich niedriger liegt als für BD. Styrol hat vermutlich auch keine höhere leukämogene Potenz als BD. Es blieb z. B. deshalb in der Auswertung von Cheng et al. als möglicher Confounder apriori unberücksichtigt.
Cheng et al. untersuchten darüber hinaus, ob die Berücksichtigung verschiedener Induktionszeiten von fünf, zehn, 15 bzw. 20 Jahren die Ergebnisse verändert. Dieses war nicht der Fall (Cheng et al, 2007), so dass die Risikoableitung - wie oben erfolgt - ohne Berücksichtigung einer Induktionsperiode durchgeführt werden kann.
Insgesamt ist zu betonen, dass sämtliche Expositionsszenarien, die in den verschiedenen Publikationen diskutiert werden, kritisch zu würdigen sind. Z. B. enthält die Publikation von Cheng verschiedene Szenarien, deren Ergebnisse sich zum Teil deutlich voneinander unterscheiden. Bei Graff lässt sich feststellen, dass der Steigungskoeffizient für Expositionsintensitäten <= 100 ppm größer ist als bei Berücksichtigung aller Expositionswerte. Dies spricht z. B. gegen eine besondere Bedeutung von Expositionsspitzen größer als 100 ppm.
Tabelle 1: Relative Rate der Leukämiemortalität in Abhängigkeit von der Kategorie der Butadien-Exposition nach der Studie von Graff et al. (2005).
| Kum. Exposition, 1,3-Butadien (BD; [ppm-J] | Langzeit-Mittelwert, 35 Jahre (a) [ppm] | Personenjahre | Leukämiemortalität | |||
|---|---|---|---|---|---|---|
| Bereich | Klassenmitte (a) | Beob. [N] | RR(1) (b)(95 Prozent-VB) | RR(2) (c)(95 Prozent-VB) | ||
| 0 | 0 | 0 | 116471 | 10 | 1 (Bezugskat.) | 1 (Bezugskat.) |
| > 0 - < 33,7 | 16,85 | 0,48 | 154443 | 17 | 1,4 (0,7-3,1) | 1,4 (0,5-3,9) |
| 33,7- < 184,7 | 109,2 | 3,12 | 144109 | 18 | 1,2 (0,6-2,7) | 0,9 (0,3-2,6) |
| 184,7- < 425 | 304,9 | 8,71 | 49411 | 18 | 2,9 (1,4-6,4) | 2,1 (0,7-6,2) |
| >= 425,0 | 600 | 17,1 | 35741 | 18 | 3,7 (1,7-8,0) | 3,0 (1,0-9,2) |
Tabelle 2: Expositions-Risiko-Beziehung für 1,3-Butadien gemäß der Ableitung des AK CM im Hinblick auf die Begründung eines Arbeitsplatzgrenzwertes (AGW).
| Butadien-Konzentration, Langzeit-Mittelwert, 35-40 Jahre Arbeitsplatzexposition | Expositionsbedingtes Lebenszeit-Leukämierisiko | |
|---|---|---|
| ppm | µg/m3 | |
| 15 | 33.660 | 3% |
| 5 | 11.220 | 1 % |
| 2 | 4.488 | 4 zu 1.000 |
| 1 | 2.244 | 2 zu 1.000 |
| 0,5 | 1.122 | 1 zu 1.000 |
| 0,05 | 112 | 1 zu 10.000 |
| 0,005 | 11 | 1 zu 100.000 |
5. Weitere zu diskutierende Aspekte
Anhand der gefundenen Expositions-Risiko-Beziehungen ist es nicht möglich, eine klare Aussage über einen von einer Linearität abweichenden Kurvenverlauf zu machen. Dies ist keine Besonderheit der Daten zu Butadien. Es ist die Regel, dass anhand epidemiologischer Untersuchungen über mögliche Zusammenhänge von Chemikalienexpositionen und Krebsrisiken keine eindeutigen Aussagen über bestimmte Kurvenverläufe der Expositions-Risiko-Beziehungen im Bereich unterhalb eines Lebenszeitrisikos von einem Prozent gemacht werden können (Roller et al, 2006).
Bei der Risikoableitung muss entschieden werden, welches Modell bzw. welches Szenario als das "realistische" bzw. am besten geeignete angesehen werden kann. Diese Ergebnisse sind für die Risikoableitung heranzuziehen. Zudem können Lebenszeitrisiken für verschiedene Szenarien berechnet werden und i. S. einer Spanne angegeben werden.
Literatur
- [1]
Ahrens, W.; Stewart, P., 2003 Retrospective exposure assessment
In: Nieuwenhuijsen, M.J., Exposure Assessment in Occupational and Environmental Epidemiology Oxford; New York: Oxford University Press, 2003
- [2]
Ahrens, W.; Behrens, T.; Mester, B.; Schmeißer, N., 2008 Epidemiologie der Arbeitswelt Bundesgesundheitsblatt (im Druck)
- [3]
Becher, H.; Steindorf, K., 1993 Epidemiologische Methoden und Wege der Risikoabschätzung Informatik, Biometrie und Epidemiologie in Medizin und Biologie, 24,1993, 14-27
- [4]
Becher, H.; Steindorf, K.; Wahrendorf, J., 1995 Epidemiologische Methoden der Risikoabschätzung für krebserzeugende Umweltstoffe mit Anwendungsbeispielen Berichte 7/95 - Forschungsbericht 116 06 089. UBA FB 95-041 Berlin: Erich Schmidt 1995
- [5]
Blair, A.; Burg, J.; Foran, J.; Gibb, H.; Greenland, S.; Morris, R.; Raabe, G.; Savitz, D.; Teta, J.; Wartenberg, D. et al., 1995 Guidelines for application of meta-analysis in environmental epidemiology. ISLI Risk Science Institute Regulatory Toxicology and Pharmacology, 22, 1995, 189-197
- [6]
Cheng, H.; Sathiakumar, N.; Graff, J.; Matthews, R.; Delzell, E., 2007 1,3-Butadiene and leukemia among synthetic rubber industry workers: exposure-response relationships Chemico-Biological Interactions, 166, 2007,15-24
- [7]
Cordier, S.; Stewart, P., 2005 Exposure Assessment In: Ahrens, W; Pigeot, I., Handbook of Epidemiology Berlin; Heidelberg; New York: Springer, 2005
- [8]
Goldbohm, R.A.; Tielemans, E.L.J.P.; Heederik, D.; Rubingh, C.M.; Dekkers, S.; Willems, M.I.; Kroese, E.D., 2006 Risk estimation for carcinogens based on epidemiological data: A structured approach, illustrated by an example on chromium Regulatory Toxicology and Pharmacology, 44, 2006, 294-310
- [9]
Graff, J.J.; Sathiakumar, N.; Macaluso, M.; Maldonado, G.; Matthews, R.; Delzell, E., 2005 Chemical exposures in the synthetic rubber industry and lymphohematopoietic Cancer mortality Journal of Occupational and Environmental Medicine, 47, 2005, 916-932
- [10]
Kromhout, H., 1994 From eyeballing to Statistical modelling: methods for assessment of occupational exposure PhD Thesis, Agricultural University of Wageningen, 1994
- [11]
Roller, M.; Akkan, Z.; Hassauer, M.; Kalberiah, F., 2006 Risikoextrapolation vom Versuchstier auf den Menschen bei Kanzerogenen Fb 1078, Bundesanstalt für Arbeitsschutz und Arbeitsmedizin, Dortmund/Berlin/ Dresden Wirtschaftsverlag NW, Bremerhaven, 2006
- [12]
Van Wijngaarden, E.; Hertz-Picciotto, I., 2004 A simple approach to performing quantitative Cancer risk assessment using published results from occupational epidemiology studies The Science of the Total Environment, 332, 2004, 81-87
10.3 Tumorlokalisationen und ihre Humanrelevanz
Es gibt eine Reihe typischer Tumorformen, die in bestimmten Nagerstämmen mit hoher teilweise auch stark variabler Spontaninzidenz auftreten und deren Relevanz für den Menschen nicht feststeht (siehe Nummer 3.1.9). Wenn deren Häufigkeit dosisabhängig gegenüber der aktuellen und der mittleren historischen Kontrolle erhöht ist, wird man in der Regel von einem expositionsbedingten Effekt sprechen. Ob man die Tumorzahlen als Ausgangsbasis für eine quantitative Risikoextrapolation heranzieht oder nicht, sollte in jedem Einzelfall dargelegt werden.
Zunächst ist zu prüfen, ob nicht auch andere Tumorformen aufgetreten sind, die keinesfalls der Spontanpathologie zugeordnet werden können und ob diese nicht bei noch niedriger Dosis und/oder in größerer Häufigkeit aufgetreten sind und allein schon aus diesem Grunde als Berechnungsgrundlage vorgezogen werden sollten.
Wesentlich ist ferner, ob es sich um eine gentoxische Substanz handelt. Bei einer gentoxischen Substanz kann eigentlich eine Humanrelevanz für kaum einen Tumortyp ausgeschlossen werden. Daher wäre in einer Default-Annahme mit dem Tumortyp zu rechnen, der die ungünstigste Risikozahl ergibt. Einzige Ausnahme hiervon wären Alpha-2u-Globulin-bedingte Nierentumoren der männlichen Ratte (siehe unten).
Ein weiterer Gesichtspunkt ist die Konzentration der Testsubstanz (oder des kritischen Metaboliten) am Zielorgan. So würde man z. B. bei einem Stoff, der seine höchsten Konzentrationen an der Eintrittspforte oder in der Niere erreicht, Tumoren an Atemwegen oder der Niere eher berücksichtigen als etwa einen endokrinen Tumor mit hoher Spontaninzidenz.
Auch bei nicht-gentoxischen Stoffen würde man solche mechanistischen Überlegungen anstellen und, falls man in solchen Fällen überhaupt eine mathematische Risikoextrapolation durchführt, nach Möglichkeit solche Tumoren auswählen, die nach Zielorgan und wirksamer Dosis zum Wirkprofil der Substanz (z. B. zytotoxisch, mitogen, endokrin) passen.
Folgende Tumorformen bei Nagetieren sind Beispiele für eine fehlende oder eingeschränkte quantitative Übertragbarkeit auf den Menschen:
Alpha-2u-Globulin-Nierentumoren der männlichen Ratte
sind ein spezies- und geschlechtsspezifisches Phänomen und können durch eine Vielfalt nicht-gentoxischer, jedoch an dieses Protein bindender Chemikalien ausgelöst werden. Dieser Effekt hat keine Relevanz für den Menschen. (IARC, 1999; siehe auch Annex 1).
Lebertumoren nach PPAR(alpha)-Rezeptor-Stimulation ("Peroxisomenproliferation")
Diese Tumoren sind in hohem Maße nagerspezifisch. Eine Relevanz für den Menschen ist in den meisten Fällen nicht gegeben. (IARC, 2000; siehe auch Annex 2).
Leukämien der Fischer-344-Ratte
Mononukleäre Leukämien sind bei Fischer-Ratten sehr häufig. Ein gehäuftes Auftreten besonders bei nicht-gentoxischen Verbindungen muss zunächst auf seine biologische Signifikanz für die Ratte beurteilt werden.
Bei einer gentoxischen Substanz kann die Humanrelevanz nicht ausgeschlossen werden. In solchen Fällen würde man allerdings prüfen, ob dies wirklich die einzige vermehrte Tumorform war, mit der man eine rechnerische Extrapolation durchführen könnte (siehe auch Annex 3).
Phäochromocytome der Fischer-344-Ratte
Mittlere historische Raten und Schwankungsbreiten sind mit einzubeziehen, ebenso die Differentialdiagnostik von der sehr häufigen altersbedingten Hyperplasie. Männliche Ratten sind offenbar gegenüber diesem Tumor eher disponiert als weibliche Ratten. Die Relevanz für den Menschen ist eingeschränkt, besonders bei nicht-gentoxischem Mechanismus und ausschließlicher Betroffenheit des männlichen Geschlechtes.
Schilddrüsentumoren bei der Ratte
Die Zufuhr von Stoffen, welchen den Glucuronidierungsweg in der Leber induzieren, können auch zu einer rascheren Elimination von Schilddrüsenhormonen aus dem Blut führen und in der Folge über den zentralen Regelkreis zu einer Stimulation des Schilddrüsengewebes (Goldstein und Tauroy, 1968; Hill et al., 1989; McClain, 1989). Leberhypertrophie oder andere Anzeichen einer generellen Enzyminduktion sind dabei nicht immer zu beobachten, wie das Beispiel tert-Butanol (NTP, 1995) zeigt, welches bei Mäusen beiderlei Geschlechter zu Schilddrüsenhyperplasien führte und bei den weiblichen Tieren vermehrt zu Adenomen. Beim Kaninchen wurde eine partielle Glucuronidierung dieses Stoffes nachgewiesen (Kamil, et al., 1953).
Beim Menschen wird in der Regel die Glucuronidierungskapazität weniger in Anspruch genommen als bei der Ratte. Außerdem sind beim Menschen T3 und T4 im Plasma hochaffin gebunden und haben eine wesentlich längere Halbwertzeit als bei der Ratte (Döhler et al., 1979; Oppenheimer, 1979; Larsen, 1982). Somit fällt für den T3/T4-Stoffwechsel des Menschen ein erhöhtes Angebot glucuronidierender Enzyme weniger ins Gewicht. Das Serum-TSH liegt ferner bei der männlichen Ratte wesentlich höher als bei der weiblichen Ratte und um ein Vielfaches höher als beim Menschen, der im Übrigen auch nicht den Geschlechtsunterschied in den TSH-Spiegeln aufweist (Chen, 1984). Die männliche Ratte ist typischerweise zu benignen und malignen Schilddrüsentumoren disponiert, während beim Menschen auch bei hoher TSH-Stimulation Schilddrüsenkarzinome nicht beobachtet werden (Refetoff et al., 1993). Somit besteht offenbar eine geringe Relevanz nichtgentoxischer Schilddrüsenkarzinogene für den Menschen. (IARC; 1999; loc. cit.).
Leydigzelltumoren
sind bei Nagern insgesamt sehr viel häufiger als beim Menschen. Ihre Relevanz für den Menschen ist gering, insbesondere dann, wenn ein Stoff nicht gentoxisch ist (Cook et al., 1999).
Lebertumoren der B6C3F1-Maus
Diese Tumoren haben eine hohe Hintergrundsrate. Nach Maronpot (1999) treten Leberadenome bei ca. 30 Prozent der männlichen Tiere und bei 15 Prozent der weiblichen Tiere auf; hepatozelluläre Karzinome bei 20 Prozent der männlichen und 10 Prozent der weiblichen Tiere. An einer quantitative Übertragbarkeit, insbesondere wenn die Substanz nicht gentoxisch ist und diese Tumorart als einzige vermehrt auftritt, bestehen Zweifel (Gamer et al., 2002).
Vormagentumoren
Bei diesen Tumoren ist die Relevanz für den Menschen wegen anderer anatomischer Verhältnisse u. U. stark eingeschränkt, insbesondere wenn es sich um nicht-gentoxische Substanzen handelt. Bei gentoxischen Substanzen ist ihre Eignung als Grundlage einer quantitativen Risikoerrechnung von Fall zu Fall zu entscheiden und hängt auch davon ab, ob noch andere Zielgewebe betroffen waren.
Annex 1 zu Anhang 10.3:
(alpha)-2u-Globuline nephropathy
is initiated by accumulation of (alpha)-2u in the phagolysosomes of the proximal convoluted tissue with subsequent acceleration of apoptosis and replicative cell turn over (Alden, 1991; Caldwell et al., 1999).
A strong association between sustained (alpha)-2u-globuline accumulation and renal neoplasia has been described by several groups of authors (Baetcke et al., 1991; Dietrich and Swenberg, 1991; IARC, 1999; Short et al., 1989; Swenberg and Lehmann-McKeeman, 1998). (alpha)-2u was shown to cause morphological transformation in the pH 6.7 SHE cell transformation assay; this effect was not achieved by other proteins nor by typical (alpha)-2u inducing Compounds such as d-li-monene or 2.2.4-trimethylpentane (Oshiro et al.).
Annex 2 zu Anhang 10.3:
PPAR(alpha)-Rezeptor-Stimulation
Bei Ratte und Maus stellt diese Form der Enzyminduktion eine potentiell lebertumordisponierende Stoffwechselsituation dar, wobei die tatsächliche Kanzerogenität der einzelnen Peroxisomenproliferatoren höchst unterschiedlich ausgeprägt ist. Von prognostischer Aussagekraft sind die Höhe der Wirkschwelle und das Ausmaß der Lebervergrößerung, weniger die maximalen Peroxisomen- und Enzymaktivitäten im Hochdosisbereich.
Nicht-Nager zeigen eine weitgehende Resistenz gegenüber dem Phänomen der Peroxisomenproliferation (siehe unten) und der hiermit assoziierten Effekte wie Enzyminduktion, Hepatomegalie und Tumorinduktion. Hamster zeigen hingegen noch schwache Effekte (Lake, 1995).
Man nimmt heute an, dass die Speziesunterschiede auf Dichte und Funktionalität eines bestimmten Rezeptortyps zurückgehen, des peroxisomenstimulierenden (PPAR(alpha))-Rezeptors, welcher bei Ratte und Maus in besonders hohem Maße und vollständiger Form exprimiert wird (Ashby et al., 1994; Bentley et al., 1993; Lee et al., 1995; Cattley et al., 1998; Maloney und Waxman, 1999). Die Stimulation der Rezeptoren führt in den Zielzellen zu einer Vielzahl von Transkriptionen bzw. Genexpressionen und morphologisch zu einer Proliferation von Zellorganellen (Peroxisomen, Mitochondrien, endoplasmatisches Retikulum), zur Suppression von Apoptose (Roberts et al., 1998) sowie zu einer zumindest initialen, bei manchen Stoffen auch kontinuierlichen Erhöhung der DNA-Synthese (Marsman et al., 1988) und Mitoserate nach Aktivierung der Kupffer'schen Sternzellen (Rose et al., 1997); die Leber ist in allen wirksamen Dosen auf längere Zeit vergrößert.
Transgene Mäuse, denen der peroxisomenstimulierende (PPAR(alpha)-)Rezeptor fehlt, zeigten mit DEHP keine Peroxisomenproliferation, keine Hepatomegalie und keine vermehrte DNA-Synthese (Ward et al, 1998). Die Bioverfügbarkeit war gegeben, dies konnte man an den Hoden- und Nierenschädigungen sehen, die allerdings schwächer ausgeprägt waren als beim Wild-Typ. Auch war selbst mit der hochwirksamen Verbindung Wy-14,643 keine Hepatokanzerogenität an PPAR(alpha)-Knock-out-Mäusen mehr erkennbar (Peters et al., 1997).
Die menschliche Leber weist 1-10 Prozent der funktionalen PPAR(alpha)-Rezeptordichte von Mäusen auf (Palmer et al., 1998). Hierin dürfte der Grund für die geringere toxikodynamische Empfindlichkeit des Menschen zu sehen sein, wie sie auch in vitro an Leberzellkulturen zum Ausdruck kommt.
Annex 3 zu Anhang 10.3:
Fischer rat leukemias
Mononuclear cell leukemia is a frequent finding in Fischer rats over 20 months old (Moloney et al., 1970; Moloney & King, 1973; Maita et al., 1987). Though rarely diagnosed up to the age of 18 months, this tumor may be the cause of up to 50 percent of all spontaneous early death cases in two years studies.
The tumor appears to originale from the spleen since splenectomized Fischer rats do not develop leukemia (Moloney & King, 1973). Historical data show spontaneous incidences from ~ 10 to 50 percent depending on the size of groups and differential diagnostic measures (Moloney et al., 1970; Coleman et al., 1977; Goodman et al., 1979; Sacksteder, 1976; Sass et al., 1975).
The disease was sometimes erroneously called monocytic leukemia or lymphoma and is correctly defined as large granulär lymphocyte (LGL) leukemia. On the basis of this more actual definition, relatively recent reviews found the following incidenes in control rats (Stromberg et al., 1983a,b; Stinson et al., 1990):
| n = 1145 | 22.2 Prozent, male | 20.5 Prozent, female |
|---|---|---|
| n = 2181 | 22.0 Prozent, male | 15.6 Prozent, female |
However, due to Variation, the incidence in smaller groups (50 rats) may range up to 50 percent and in such cases represent a cluster.
The pattern of morphologic, immunologic, biochemical and functional characteristics of the LCL cells resembles those of normal large granulär lymphocytes and in some respects also NK cells (Ward & Reynolds, 1983; Reynolds et al., 1981; Stromberg et al., 1983a, b). The tumor is tranplantable (- however, not with cell free lysates) and, after transplantation, causes all clinical and immunological features observed also after spontaneous occurrence (Reynolds et al., 1984; Stromberg et al., 1985). So far, there is no evidence for a viral etiology.
A considerable number of genotoxic and non-genotoxic chemicals was associated with an increased incidence of this tumor.
Examples are:
| NTP bioassay program | non-NTP studies | ||
|---|---|---|---|
| - | 2-Amino-5-nitrothiazol | - | Ethylene Oxide |
| - | 3,3'-Dimethoxy-benzidine4.4-diisocyanate | ||
| - | Arocolor 1254 | ||
| - | 2.4.6.-TCP | ||
| - | Phenol | ||
| - | Sulfisoxazole | ||
| - | Pyridine | ||
| - | Piperonylbutoxide | ||
| - | Lasiocarpine | ||
| - | Dimethylmorpholino-phosphoramidate | ||
| - | Diazinone | ||
| - | Ally(l)thiocyanate | ||
| - | Ally(l)isovalerate | ||
| - | Diallylphthalate | - | Ethylene Glycol (males) |
| - | Butylbenzylphthalate | - | DINP |
| - | (DEHP) | - | Sanitizer 900 |
This shows that many Compounds associated with increased LGL leukemia were non-genotoxic.
Other Compounds have shown reduced LGL leukemia incidence, e.g.:
| NTP bioassay program: | |
|---|---|
| - | 1.1-Aminoundecanoic acid |
| - | 2-Biphenylamine |
| - | CI-Disperse yellow |
| - | CI-Solvent yellow |
| - | CI-Acid orange |
| - | D & C red 9 |
| - | Propylgallate |
| - | Monuron |
| - | Ethoxyethanol |
A review in 1983 described correlations between decreased incidence of leukemia and elevated incidence of liver tumor (Haseman, 1983). Over the past decades, there is a general trend for an increase in leukemias rates among male F344 rats in NCI/NTP studies. This is possibly related to the higher body weights in more recent studies (Haseman et al., 1989).
Conclusion:
LGL leukemia is a typical and frequent tumor in ageing Fischer rats. The etiology so far is unknown. Many Compounds that were associated with an increased occurrence of LCL cells leukemia did not show genotoxicity. Quite frequently, it was the only increased tumor incidence that was observed in the course of a 2-year bioassay, either with or without dose relation. Furthermore, the spontaneous incidence within a 50 rat collective may be highly variable (-> cluster formation). For those reasons an increased incidence of LCL cell leukemia is not regarded as a sufficient criterium to define a substance as carcinogenic. A more recent review by Caldwell (1999) comes to similar conclusions.
Literatur
- [1]
Alden, C.L., 1991 Urinary System In: Haschek, W.M.; Rousseaux, C., Handbook of Toxicologic Pathology. Academic Press Inc., San Diego, 1991, 315-387
- [2]
Ashby, J., Brady, A., Elcombe, CR., Elliott, B.M., Ishmael, J., Odium, J. Tugged, J.D., Kettle, S., Purchase, I.F.H., 1994 Mechanistically-based human hazard assessment of peroxisome proliferator-induced hepato-carcinogenesis Hum. Exp. Toxicol., 13, Suppl. 2, 1994, 1-117
- [3]
Baetcke, K.P.; Hard, G.C.; Rodgers, LS.; McGaughy; R.E.; Tahan, L.M., 1991 Alpha2u-globulin: Association with chemically induced renal toxicity and neoplasia in the male rat EPA/625/3-91/019F, Risk Assessment Forum, U.S. Environmental Protection Agency, Washington, DC, 1991
- [4]
Bentley, P., Calder, I., Elcombe, C., Grasso, P., Stringer, D., Wiegand, HJ., 1993 Hepatic peroxisome proliferation in rodents and its significance for humans Food Chem. Toxicol, 31, 1993, 857-907
- [5]
Caldwell, DJ., 1999 Review of mononuclear cell leukemia in F-344 rat bioassays and its significance to human cancer risk: a case study using alkyl phthalates Regulatory Toxicology and Pharmacology, 30, 1999, 45-53
- [6]
Caldwell, DJ.; Eldridge, S.R.; Lington, A.W.; McKee, R.H., 1999 Retrospective evaluation of alpha 2u-globulin accumulation in male rat kidneys following high doses of diisononyl phthalate Toxicological Sciences, 51, 1999, 153-160
- [7]
Cattley, R.C., DeLuca, J., Elcombe, C., Fenner-Crisp, P., Lake, B.G., Marsman, D.S., Pastoor, TA., Popp, J.A., Robinson, D.E., Schweiz, B., Tugwood, J., Wahli, W., 1998 Do peroxisome proliferating Compounds pose a hepatocarcinogenic hazard to humans? Regulatory Toxicology and Pharmacology, 27, 1998, 47-60
- [8]
Chen, H.J., 1984 Age and sex difference in serum and pituitary thyrotropin concentrations in the rat: Influence by pituitary adenoma Toxicol. Pathol., 17,1984, 266-293
- [9]
Coleman, G.L.; Barthold, S.W.; Osbaldistan, G.W; Foster, S.J.; Jonas, A.M., 1977 Pathological changes during aging in barrier-reared Fischer 344 male rats Journal of Gerontology, 32,1977, 258-278
- [10]
Cook, J.C., Klinefelter, G.R., Hardisty, J.F., Sharpe, R.M., Foster, P.M.D., 1999 Rodent Leydig Cell Tumo-rigenesis: A Review of the Physiology, Pathology, Me-chanisms, and Relevance to Humans Crit. Rev. Toxicol., 29,1999, 169-261
- [11]
Dietrich, D.R.; Swenberg, J.A., 199.1 The presence of (alpha)2u-globulin is necessary for d-limonene of male rat kidney tumors Cancer Research, 51, 1991, 3512-3521
- [12]
Döhler, K.D., Wong, C.C., von zur Mühlen, A., 1979 The rat as a model for the study of drug effects on thyroid function: consideration of methodological problems Pharmacol. Ther., 5, 1979, 305-318
- [13]
Gamer, A.O., Jäckh, R., Leibold, E., Kaufmann, W., Gembardt, Chr., Bahnemann, R., van Ravenzwaay B. (2002) Investigations on cell proliferation and enzyme induction in male rat kidney and female mouse liver by Tetrahydrofurane Toxicol. Sci., 70, 2002, 140-149
- [14]
Goldstein, J.A., Tauroy, A., 1968 Enhanced exeretion of thyroxine glucuronide in rats pretreated with benzpyrene Biochem. Pharmacol., 17, 1968, 1044-1065
- [15]
Goldsworthy, TL.; Monticello, TM.; Morgan, K.T.; Bermudez, E.; Wilson, D.M.; Jäckh, R.; Butterworth, B.E., 1991 Examination of potential mechanisms of carcinogenicity of 1,4-dioxane in rat nasal epithelial cells and hepatocytes Archives of Toxicology, 65, 1991, 1-9
- [16]
Goodman, D.G.; Ward, J.M.; Squire, R.A.; Chu, K.C.; Linhart, M.S., 1979 Neoplastic and nonneoplastic lesions in aging F344 rats Toxicology and Applied Pharmacology, 48, 1979, 237-248
- [17]
Haseman, J.K., 1983 Patterns of tumor ineidence in two-year cancer bioassay feeding studies in Fischer 344 rats Fundamental and Applied Toxicology, 3,1983,1-9
- [18]
Haseman, J.K.; Huff, J.E.; Rao, G.N.; Eustis, S.L., 1989 Sources of variability in rodent carcinogenicity studies Fundamental and Applied Toxicology, 12, 1989, 793-804
- [19]
Hill, R.N., Erdreich, L.S., Paynter, O.E., Roberts, P.A., Rosenthal, S.L., Wilkinson, C.F, 1989 Thyroid follicular cell carcinogenesis Fundam. Appl. Toxicol., 12, 1989, 629-697
- [20]
IARC, International Agency for Research on Cancer, 1999 IARC Scientific Publication No. 147. Species Differences in Thyroid, Kidney and Urinary Bladder Carcinogenesis IARC, International Agency for Research on Cancer, Lyon, 1999
- [21]
IARC, International Agency for Research on Cancer, 2000 IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Vol. 77. Some Industrial Chemicals WHO, World Health Organization, Geneva, 2000
- [22]
Kamil, I.A., Smith, J.N., Williams, R.T., 1953 Studies in detoxication. The metabolism of aliphatic alcohols. The glucuronic aeid conjugation of aeyclic aliphatic alcohols Biochem. J., 53, 1953, 129-136
- [23]
Lake, B.G., 1995 Mechanisms of hepatocarcinogenicity of peroxisome-proliferatirig drugs and chemicals Ann. Rev. Pharmacol. Toxicol, 35, 1995, 483-507
- [24]
Larsen, P.R., 1982 The thyroid In Wyhgaarden, JB. & Smith, L.H. eds. Cecil Textbook of Medicine. Philadelphia: Saunders. 16th ed., 1201-1225
- [25]
Lee, S.S., Pineau, T, Drago, J., Lee, E.J., Owens, J.W., Kroetzz, D.L., Femandez-Salguero, P.M., Westphal, H, Gonzales, F.J., 1995 Targeted disruption of the alpha isoform of the peroxisome proliferator-activated re-ceptor gene in mice results in abolishment of the pleio-tropic effects of peroxisome proliferators Mol. Cell. Biol., 15,1995, 3012-3022
- [26]
Maita, K.; Hirano, M.; Harada, T; Mitsumori, K.; Yoshida, A.; Takahashi, K.; Nakashima, N.; Kitazawa, T.; Enomoto, A.; Inui, K.; Shirasu, Y., 1987 Spontaneous tumors in F344/DuCrj rats from 12 control groups of chronic and oncogenicity studies Journal of Toxicological Sciences, 12, 1987, 111-126
- [27]
Maloney, E.K. and Waxman, D.J., 1999 Trans-Activation of PPARá and PPAR[gamma] by Structurally Diverse Environmental Chemicals Toxicol. Appl. Pharmacol., 161,1999,209-218
- [28]
Maronpot, R.R., 1999 Pathology of the mouse: reference and atlas 1st edition, Cache River Press, Saint Louis, 1999
- [29]
Marsman, D.S., Cattley, R.C., Conway, J.G., Popp, J.A., 1988 Relationship of hepatic peroxisome proliferation and replicative DNA synthesis to the hepatocarcinogenicity of the peroxisome proliferators, di(2-ethylhexyl)phthalate and [4-chloro-6-(2,3,-xylidino)-2-pyrimidini-[thio]acetic aeid (Wy-14,643) in rats Cancer Res., 48,1988, 6739-6744
- [30]
Matthews, E.J.; Spalding, J.W; Tennant, R.W, 1993 Transformation of BALB/c-3T3 cells: V. Transformation responses of 168 chemicals compared with mutagenicity in Salmonella and carcinogenicity in rodent bioassays Environmental Health Perspectives, 101, Suppl. 2,1993, 347-482, as cited in SIDS Profile (1996)
- [31]
McClain, R.M., 1989 The significance of hepatic mi-crosomal enzyme induction and altered thyroid function in rats: Implications for thyroid gland neoplasia Toxicol. Pathol., 17,1989, 294-306
- [32]
Miyagawa, M.; Katsuta, O.; Tsuchitani, M.; Yoshikawa, K., 1997 Measurement of replicate DNA synthesis (RDS) by a 5-bromo-2'-deoxyuridine (BrdU) labeling technique for detection of hepatocyte proliferation Journal of Veterinary Medical Science, 59,1997, 45-49
- [33]
Moloney, W.C.; Boschetti, A.E.; King, V.P., 1970 Spontaneous leukemia in Fischer rats Cancer Research, 30, 1970,41-43
- [34]
Moloney, W.C.; King, V.P., 1973 Reduction of leukemia ineidence following splenectomy in the rat Cancer Research, 33,1973, 573-574
- [35]
Mortelmans, K.; Haworth, S.; Lawlor, T; Speck, W; Tainer, B.; Zeiger, E., 1986 Salmonella mutagenicity tests: II. Results from the testing of 270 chemicals Environmental Mutagenesis, 8, Suppl. 7, 1986, 1-119, as cited in NTP Technical Report on the Toxicology and Carcinogenesis Studies of Tetrahydrofuran (CAS No. 109-99-9) in F344/n Rats and B6C3F1 Mice (Inhalation Studies), NIH Publication No. 96-3965 (1996)
- [36]
NTP, National Toxicology Program, 1995 Toxicology and Carcinogenesis Studies of t-Butyl Alcohol in F344/ N Rats and B6C3F1 Mice (Drinking Water Studies). TR 436 U.S. Department of Health and Human Services; Public Health Service, 1995
- [37]
Oppenheimer, J.H., 1979 Thyroid hormone action at the cellular level Science, 203,1979, 971-979
- [38]
Oshiro, Y.; Alden, C.L., 1998 Exploration of the transformation potential of a unique male rat protein alpha2u-globulin using hamster embryonic cells Toxicologic Pathology, 26, 1998, 381-387
- [39]
Palmer, C.A.N., Hsu, M.H., Griffin, K.J., Raucy, J.L., Johnson, E.F., 1998 Peroxisome proliferator activated receptor-(alpha) expression in human liver Mol. Pharmacol. 53, 1998,14-22
- [40]
Peters, J.M., Cattley, R.C., and Conzalez, F.J., 1997 Role of PPAR alpha in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643 Carcinogenesis, 18, 1997, 2029-2033
- [41]
Refetoff, S. Weiss, R.E. & Usala, S.J., 1993 The Syndromes of resistance to thyroid hormone Endocrine Rev., 14, 1993, 348-399
- [42]
Reynolds, C.W; Timonen, T; Herberman, R.B., 1981 Natural killer (NK) cell activity in the rat. I. Isolation and characterization of the effector cells Journal of Immunology, 127,1981, 282-287
- [43]
Reynolds, C.W; Bere, E.W.; Ward, J.M., 1984 Natural killer activity in the rat. III. Characterization of trans-plantable large granulär lymphocyte (LGL) leukemias in the F344 rat Journal of Immunology, 132,1984, 534-540
- [44]
Roberts, R.A., James, N.H. Woodyatt, HJ., Macdonald, N. Tugwood, J.D., 1998 Evidence for the suppression of apoptosis by the peroxisome proliferator activated receptor alpha (PPAR alpha) Carcinogenesis, 19,1998,43-48
- [45]
Rose, M.L., Germolec, D.R., Schoonhoven, R., Thurman, R.G., 1997 Kupffer cells are causally responsible for the mitogenic effect of peroxisome proliferators Carcinogenesis, 18,1997,1453-1456
- [46]
Sass, B., Rabstein, L.S., Madison, R. et al., 1975 Incidence of spontaneous neoplasms in F344 rats throughout natural life-span J. Nat. Canc. Inst., 54, 1975, 1449-1456
- [47]
Schulte-Hermann, R., 1983 Promotion of spontaneous preneoplastic cells in rat liver as a possible explanation of tumor production by nongenotoxic Compounds Cancer Research, 43,1983, 839-844
- [48]
Short, B.G.; Burnett, V.L.; Swenberg, J. A., 1989 Elevated proliferation of proximal tubule cells and localization of accumulated alpha 2u-globulin in F344 rats during chronic exposure to unleaded gasoline or 2,2,4-trimethylpentane Toxicology and Applied Pharmacology, 101,1989,414-431
- [49]
Stinson, S.F; Schuller, H.M.; Reznik, G., 1990 Atlas of tumor pathology of the Fischer rat CRC Press Inc., Boca Raton, Florida, 1990
- [50]
Stromberg, P.C.; Vogtsberger, L.M.; Marsh, L.R.; Wilson, F.D., 1983a Pathology of the mononuclear cell leukemia of Fischer rats. IL Hematology Veterinary Pathology, 20, 1983, 709-717
- [51]
Stromberg, P.C.; Vogtsberger, L.M.; Marsh, L.R., 1983b Pathology of the mononuclear cell leukemia of Fischer rats. III. Clinical chemistry Veterinary Pathology, 20, 1983, 718-726
- [52]
Stromberg, P.C.; Vogtsberger, L.M.; McMurray, D.N.; Marsh, L.R.; Kotur, M.S.; Brown, CA., 1985 Behavior of transplanted large granulär lymphocyte leukemia in Fischer 344 rats Laboratory Investigation, 53, 1985, 200-208
- [53]
Swenberg, J.A.; Lehman-McKeeman, L.D., 1998 á2u-Globulin associated nephropathy as a mechanism of renal tubulär cell carcinogenesis in male rats In: Species Differences in Thyroid, Kidney, and Urinary Bladder Carcinogenesis, IARC Scientific Publications No. 147. International Agency for Research on Cancer, Lyon, 1998
- [54]
Ward, J.M.; Reynolds, C.W, 1983 Large granulär lymphocyte leukemia. A heterogeneous lymphocytic leukemia in F344 rats American Journal of Pathology, 111, 1983,1-10
- [55]
Ward, J.M., Peters, J.M., Perella, C.M., Gonzalez, F.J., 1998 Receptor and Nonreceptor-Mediated Organ-Specific Toxicity of Di(2-ethylhexyl) phthalate (DEHP) in Peroxisome Proliferator-Activated Receptor (alpha)-Null Mice Toxicol. Pathol., 26,1998, 240-246
- [56]
Yamazaki, K.; Ohno, H.; Asakura, M.; Narumi, A.; Ohbayashi, H.; Fujita, H.; Ohnishi, M.; Katagiri, T; Senoh, H. et al., 1994 Two-year Toxicological and Carcinogenesis Studies of 1,4-Dioxane in F344 Rats and BDF1 Mice - drinking studies Japan Bioassay Laboratory, Proceedings - Second Asia-Pacific Symposium on Environmental and Occupational Health, 22-24 July 1993, Kobe University, National University of Singapore, 193-198